
Interpreting the Microwave Spectra of Diatomic Molecules

Department of Chemistry, Saint Mary’s University, Halifax, NS B3H 3C3, Canada
Spectrosc. J. 2023, 1(1), 3-27; https://doi.org/10.3390/spectroscj1010002
Submission received: 3 January 2023
/
Revised: 1 February 2023
/
Accepted: 1 February 2023
/
Published: 8 February 2023
(This article belongs to the Special Issue Feature Papers in Spectroscopy Journal)
Abstract
:A brief review of the theory of the rigid rotor and its application to microwave spectroscopy is given. By careful selection of examples, procedures are given for the analysis of successively more complicated spectra, and the theory is extended to the harmonic nonrigid rotor and anharmonic nonrigid rotor when needed. The microwave spectra of carbon monoxide, and of some alkali halides, provide excellent examples for analysis and for student exercises.
1. Introduction
Microwave radiation corresponds to the frequency range from approximately 0.3 to 300 GHz and is important in modern satellite and cell phone communication. Microwaves lie between radio waves of lower frequency and infrared radiation of higher frequency and have a wavelength from 0.001 to 1 m [1]. Microwave spectroscopy gives information about the dipole moment and moments of inertia of a gas-phase molecule because the photon energies correspond to transitions between different rotational levels [2]. Radio telescopes can measure this radiation in space, supporting accurate estimates of the age of the universe through the cosmic microwave background, detecting the presence of small molecules in cosmic dust clouds, and inferring the presence of dark matter by measuring the rotational speed of galaxies via the hydrogen line at 21 cm. On Earth, microwave spectrometers have made their way into a few undergraduate laboratories [3,4], as have specialized computer programs for their prediction and analysis [5,6,7,8]. In some cases, Fourier-transform infrared spectrometers can be used to observe rotational bands [9]. The simplest spectra to analyze are molecules containing only two atoms. If performed correctly, very accurate bond distances, devoid of solvent or crystal packing effects, can be calculated.
2. Basic Theory
A diatomic molecule can be regarded as two masses, m1 and m2, separated by a distance r. If we assume that the distance r is fixed, then we refer to this as the rigid rotor approximation. This problem can be shown to be equivalent to a single mass μ rotating at the same distance r about an origin located at the center of mass of the molecule. This reduced mass μ is given as follows:
The moment of inertia of this system, I, is given by
The Schrödinger equation, applied to this system, gives the well-known energy levels [10]:
where J is the rotational quantum number, and B is the rotational constant, given by
where h is Planck’s constant. The solution of the Schrödinger equation is rather tedious and is skipped in many texts [11]. The energy levels are 2J + 1-fold degenerate, corresponding to different values of a second quantum number m. The quantum numbers J and m are analogous to l and ml discussed in the solution to the hydrogen atom, since the solution is the same. Because the selection rule for rotational transitions [12,13,14] is , and because we normally deal with absorption (J corresponds to the lower state), the transitions are observed at
A series of equally spaced lines, separated by 2B, should be observed. The rotational constant is often reported as a frequency (B/h, Hz) or as a wavenumber (B/hc, cm−1). We shall refer to this simplest possible model (Equation (3)) as Model 0. Because B depends on the moment of inertia, and thus the mass of the atoms, different isotopologues will have different values of the rotational constant.
The above treatment is the deepest extent to which many texts treat the rotational spectroscopy of a diatomic molecule [15,16,17,18]. This simple approach does work for some diatomic molecules in which only the lowest vibrational level is significantly populated. We shall see below that the centrifugal distortion term and the vibration–rotation interaction are also needed to understand other spectra. Other texts acknowledge the existence of one [19,20] or both [10,21,22] of these terms, but without derivation. We provide these derivations below.
3. Materials and Methods
Zielinski advocates for the recreation of the laws of nature “through discovery and exploration the order that we see” [23]. We concur with this approach. The aim below is to present initially very simple spectra from the literature, followed by progressively more complicated spectra, in order to introduce concepts and tools on an as-needed basis. Microsoft Excel [24] with Solver [25] was used to analyze the spectra in each case, with the spreadsheets given as Supporting Information.
4. Results
4.1. Carbon Monoxide (CO)
The microwave spectrum of carbon monoxide was measured by Gilliam, Johnson, and Gordy [26]. Transitions were observed at 115,270.56 ± 0.25 MHz for 12C16O and at 110,201.1 ± 0.4 MHz for 13C16O (isotopically enriched to 14% 13C). The X-ray crystal structure of carbon monoxide gives a distance of 1.0629 Å [27,28]. Using this distance, the predicted value of 2B of the lighter isotopologue is 130,500 MHz (see Supplementary Material, Excel file CO.xlsx). The agreement between the predicted value of 2B and the observed value (to within 15%) suggests that the observed transition corresponds to J = 0→J = 1. This illustrates one method for assigning J, which is normally the first step. If we have two or more isotopologues, then we can check the isotopic assignments by comparing the ratio of the rotational constants to the inverse of the ratio of the reduced masses, assuming the bond length of the isotopologues is the same. In this case, B12/B13 = 1.04600, whereas μ13/μ12 = 1.04612. The agreement is reasonable. The bond distances are calculated to be 1.130895 Å (12C16O) and 1.130832 Å (13C16O). These differ from each other slightly because they correspond to the first vibrational state r0, and they differ from those reported in ref. [26] because of the slightly smaller value of Planck’s constant used therein. Carbon monoxide has a longer bond length in the gas phase than in the solid.
The two pedagogical goals achieved here are (1) demonstrating the assignment of rotational quantum number J using extraspectroscopic information (XRD), and (2) confirming isotopologue assignment by comparing B ratios to μ ratios.
4.2. Alkali Halides
The alkali metal to halogen distances in the alkali halides in both the crystal and gas-phase, as measured by X-ray [29] and/or electron diffraction [30], respectively, are given in Table 1. Even though the crystal measurements are at a lower temperature, the distances are longer than the gas phase by an average of 12% for the NaCl-like structures and 17% for the CsCl-like structures. The major reason is that in the gas phase, the atoms only have one nearest neighbor, whereas in the solid, the atoms have six (NaCl-like) or eight (CsCl-like) equidistant nearest neighbors. The missing gas-phase results can be estimated for the purposes of predicting the rotational constants if needed.
4.3. Cesium Iodide (CsI)
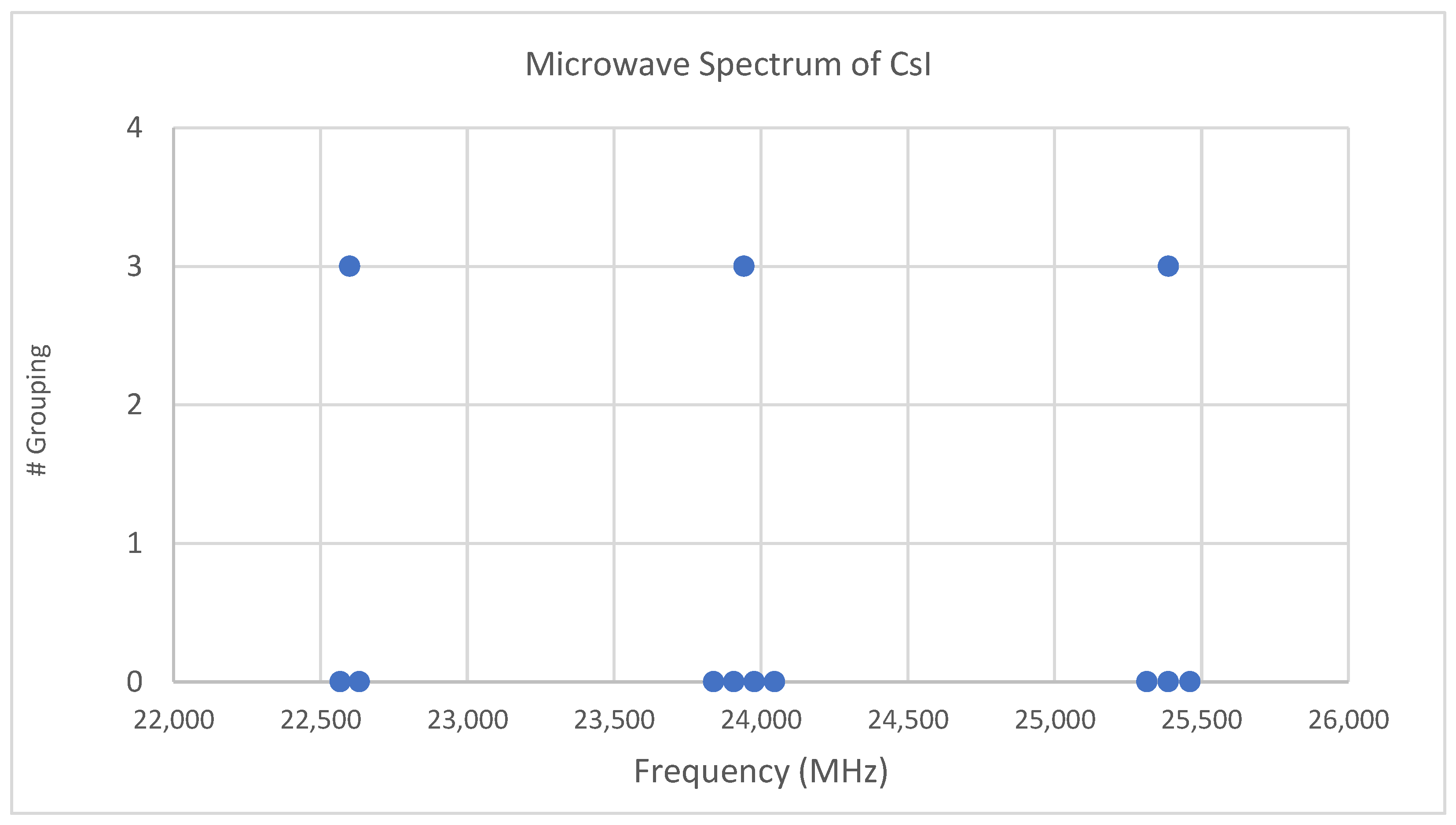
Both cesium and iodine are monoisotopic, so complications from multiple isotopologues are avoided. The predicted value of 2B from the electron diffraction value is 1340 MHz (see Supplementary Material, Excel file CsI.xlsx). The spectrum of cesium iodide in the region 22–26 GHz was measured [31] at 640 °C, and the frequencies observed (error 0.1 MHz) are given in Figure 1. Inspection of the nine frequencies (n = 9) demonstrates that there appear to be three major groupings (clusters) of frequencies. One-dimensional cluster analysis has recently been applied by the author to confirm this [32]. This tells us that the primary variation in the spectra is due to three distinct values of an independent variable, which we identify as J.
We now turn to determining J for these clusters. The cluster averages (MHz) for N = 3 are given as 22,600, 23,942, and 25,387, with differences of 1343 and 1445 (average 1394 ± 51). The predicted value of 2B is 1340 MHz, and it is therefore reasonable to assume that these clusters are due to successive values of J. The differences between the highest frequency datum (head) of each cluster are 1414.14 and 1414.13 MHz. Dividing the k = 3 cluster averages by 1340 MHz gives 16.87, 17.87, and 18.94, which suggests that these bands should be assigned as J = 16, 17, and 18, respectively. However, dividing the cluster averages by the average separation between clusters gives 16.21, 17.18, and 18.22, suggesting J = 15, 16, and 17, respectively. Dividing the heads by the head difference gives 16.00431, 17.00432, and 18.00431, which would suggest that this is the most accurate way to estimate the correct values of J + 1 (J = 15, 16, and 17).
The two pedagogical goals achieved here are (1) introducing the utility of cluster analysis, and (2) if more than one value if J is believed to be present, comparing the assignment of J using cluster averages/predicted 2B (extraspectroscopic), cluster averages/cluster differences, and head/head difference (preferred).
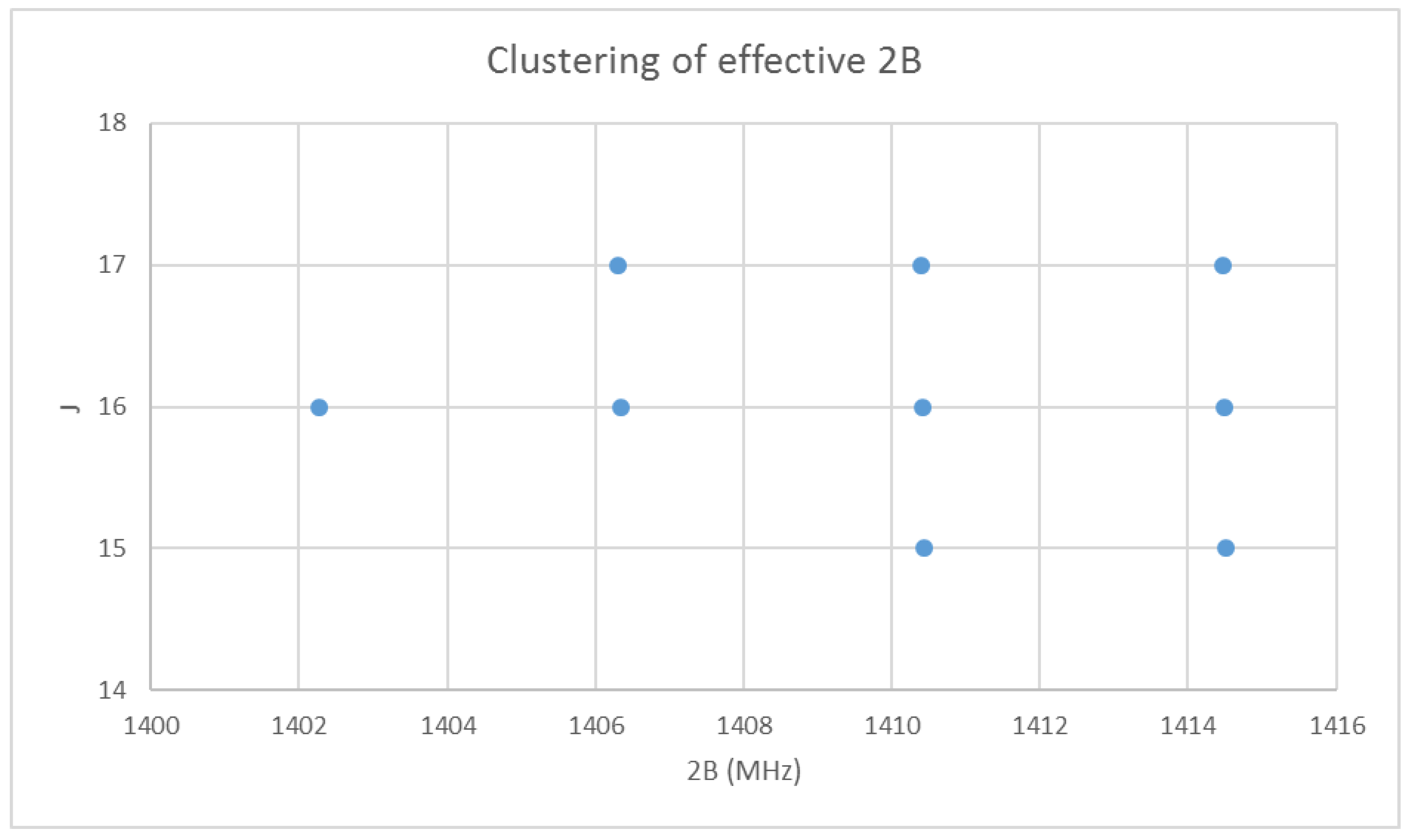
With the rotational quantum numbers J assigned, we may now calculate the effective rotational constants for each point of the spectrum. These are shown in Figure 2. The variation within each frequency cluster (or effective rotational constants) is not yet explained; however, it appears quite regular. Cluster analysis may also be carried on the effective rotational constants to show that there are four clusters [32]. There is also a very slight decrease in effective rotational constant with increasing J. We must return to the theory to explain this.
5. Advanced Theory
A diatomic molecule can vibrate as well as rotate. If the oscillation is harmonic, then for small oscillations x about the internuclear distance re, the Schrödinger equation is
where k is the spring constant. If we scale x by the constant α1/2 to give the dimensionless ξ, where
then the solutions to Equation (6) are a product of a Gaussian and a Hermite polynomial Hv:
where v is the vibrational quantum number [33]. The energy levels are given by
where . The energy levels of the molecule as a whole (ignoring electronic states) are the sum of the vibrational and rotational terms:
If the molecule is not a harmonic oscillator, but still obeys the Born–Oppenheimer approximation, then the potential can be written as a general function . The Schrödinger equation can still be separated into rotational and vibrational components, and the solutions to the rotational component are still the familiar spherical harmonics. The radial equation becomes
The last term is related to the potential energy associated with a “centrifugal” force [34] due to angular momentum J. With the substitution
Equation (11) may be simplified to
If we consider a harmonic oscillator
then we may transform the independent variable
and Equation (13) becomes
If x is small and can be ignored in the denominator of the last term, then Equation (16) becomes
This is simply the harmonic oscillator equation with the rotational energy separated out, and the energy will simply be the sum of the rotational and vibrational terms:
We note that the vibrational frequency νe and rotational constant Be correspond to the minimum in the potential energy (the subscript e refers to equilibrium).
What happens if x becomes too large to ignore? In this case, Equation (16) is equivalent to
which can be approximated, as long as , as
One way to solve this problem is by using perturbation theory on the zeroth-order harmonic oscillator equation, which we will discuss later. We examine an alternate approach. If the infinite series is truncated after the linear or quadratic terms, we obtain
and
Both of these equations are exactly soluble, because the effective potential is still quadratic. Equation (21) becomes (after rearranging and completing the square)
If we substitute
then we obtain
This is just the normal harmonic oscillator equation with usual solution, except an extra term is taken out, which is formally identified as the centrifugal distortion term [35]:
The harmonic oscillator solutions are no longer centered about x = 0, but increase with rotational constant by δ1.
If we now include the quadratic term as well, Equation (22) becomes
Expanding and collecting terms, we obtain
If we let
then
If we substitute
then we obtain
Some third- and fourth-order (in ) pure rotational terms have been subtracted out, and we are left with the usual harmonic oscillator problem, but with a different force constant k′. The main effect is on the vibrational energy levels:
and, therefore, this effect is called the vibration–rotation interaction. It can be approximated as
We may let
When combined with the significant rotational terms, we obtain
If we let the effective rotational constant for vibrational level v be
then we can see that each vibrational level will have a different effective rotational constant, and that the effective rotational constants are approximately evenly spaced. [36] Although Equation (35) predicts that αe is positive, in practice it is negative because of anharmonicity (below). The variation in the effective rotational constant, as seen in Figure 2, is thus explained as being due to the vibrational quantum number v.
Alternatively, we may apply perturbation theory to the harmonic oscillator, with the following perturbation:
First-order perturbation theory will have no contribution from terms of odd order in x. The first-order correction to the energy (from terms of even order in x) is
where f is a quadratic function in . The second-order correction to the energy contains the centrifugal distortion correction (from the linear term in x), a vibrational anharmonicity term involving , and additional terms involving [34,37]. The j = 1 term here is usually larger than the corresponding first-order term and is opposite in sign, explaining why αe is usually negative. The first-order correction involving the quadratic function f and the j = 2 term of the second-order correction can be related to γe below.
The pedagogical goal achieved here is to show what happens to the energy levels when the molecule cannot be treated as a harmonic oscillator and rigid rotor, in order to explain some of the finer structure in the spectra of alkali halides.
6. More Results
6.1. Cesium Iodide (CsI, Reprise)
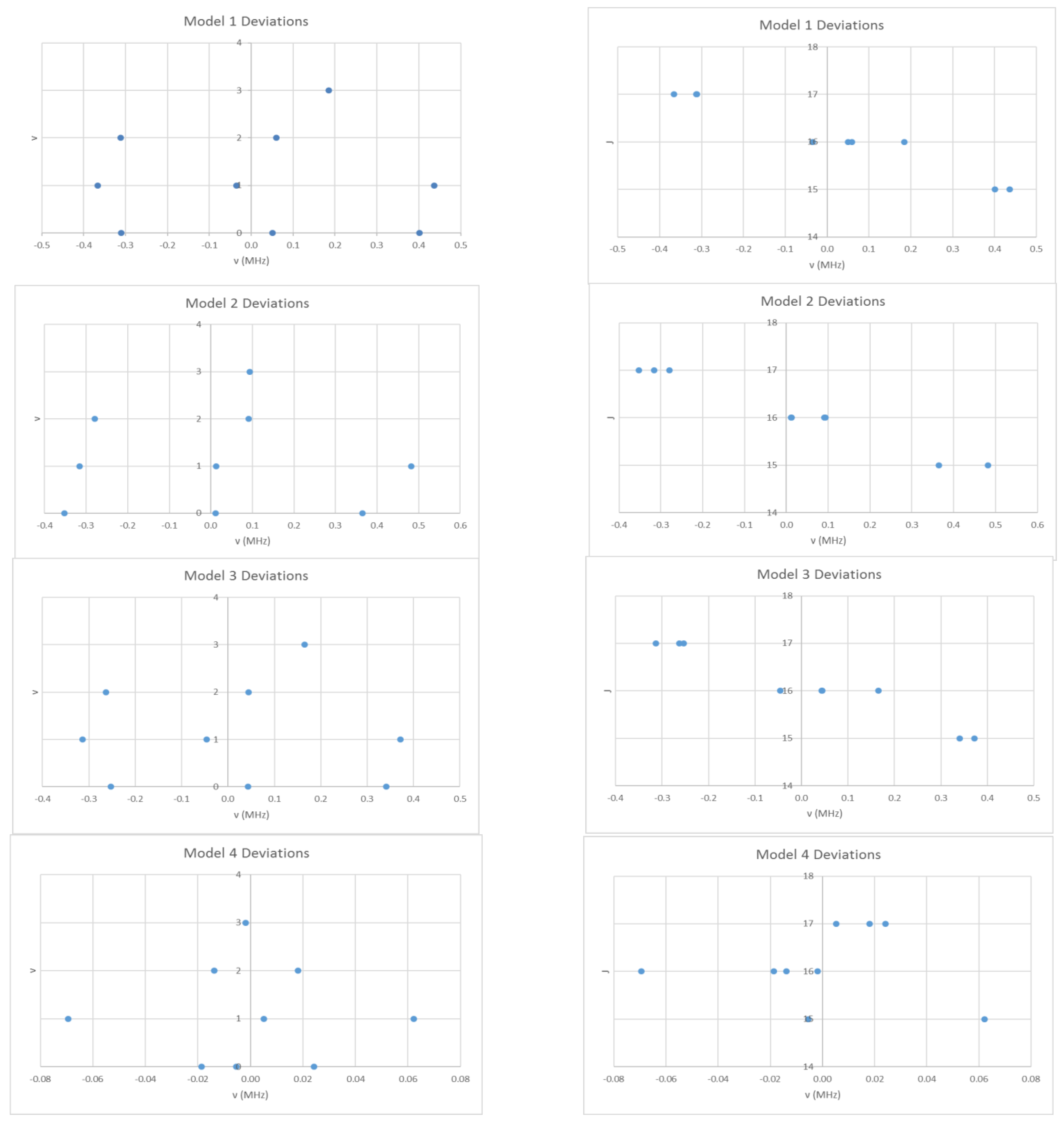
Returning to Figure 2, it is now clear that the four clusters represent different vibrational states, and we assign them to , with the highest frequency corresponding to . We may either model the effective rotational constants via Equation (37), or model the rotation spectra itself using Model 1:
These will give slightly different results under equal weighting least squares. We obtain Be = 708.266 and αe = 2.040 MHz, sum of squares error (SSE) = 0.7213 (Model 1, Table 2). In Figure 3, the error in this model is plotted as a function of the quantum numbers v and J. The error is up to five times the estimated error (0.1 MHz) in each individual measurement, which suggests that improvements can be made to the model. In addition, the error appears to have a quadratic dependence on v and a linear dependence on J. Model 1 may be improved by adding a quadratic term in v + ½ to give Model 2:
or by adding a centrifugal distortion term to give Model 3:
Model 2 removes the quadratic dependence of the error on v, (Be = 708.269, αe = 2.045, γe = 0.0015 MHz, SSE = 0.6840), whereas Model 3 keeps it (Be = 708.280, αe = 2.040, De = 2.57 × 10−5 MHz, SSE = 0.5194). The error is reduced to an acceptable level only by including both terms (Model 4):
which gives a much lower SSE (Be = 708.362, αe = 2.043, γe = 0.0011, De = 1.62 × 10−4 MHz, SSE = 0.0102). The estimated Cs-I bond distance is 3.3151 Å, which is about 0.1 Å shorter than the electron diffraction result. This microwave result corresponds to re, whereas the electron diffraction result would be a Boltzmann average over many vibrational and rotational states. Even so, each unit increase in vibrational quantum number adds only about 0.005 Å to rv for CsI.
The pedagogical goals achieved here show that for CsI, (1) the most important extension to the simplest model is to include the vibrational number dependence of the effective rotational constants via Model 1, (2) they demonstrate how to assign vibrational quantum numbers, and (3) the incorporation of either the De and γe terms lead to an approximately equal (but small) reduction in error, whereas incorporation of both leads to a much better fit.
6.2. Cesium Bromide (CsBr)
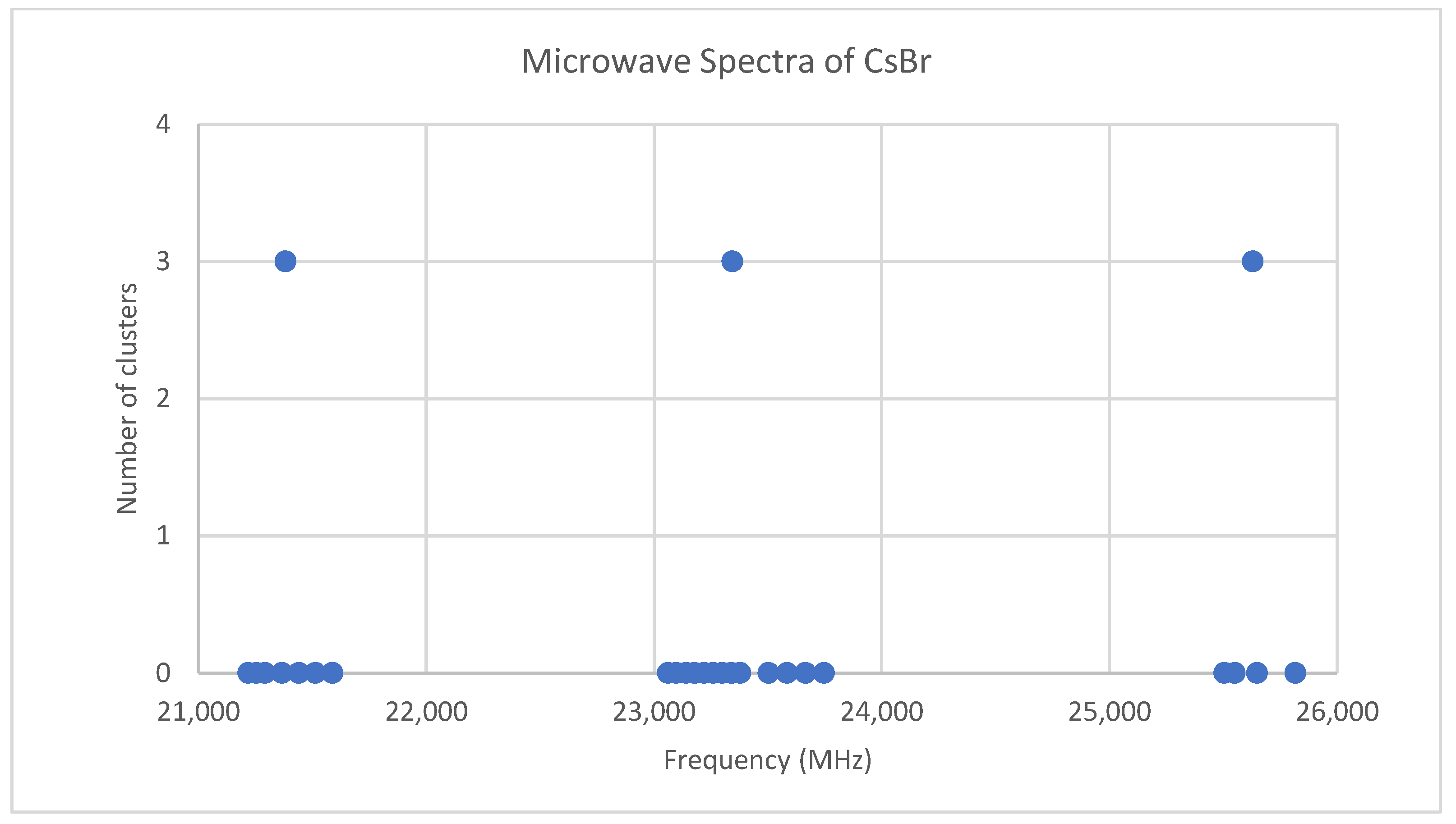
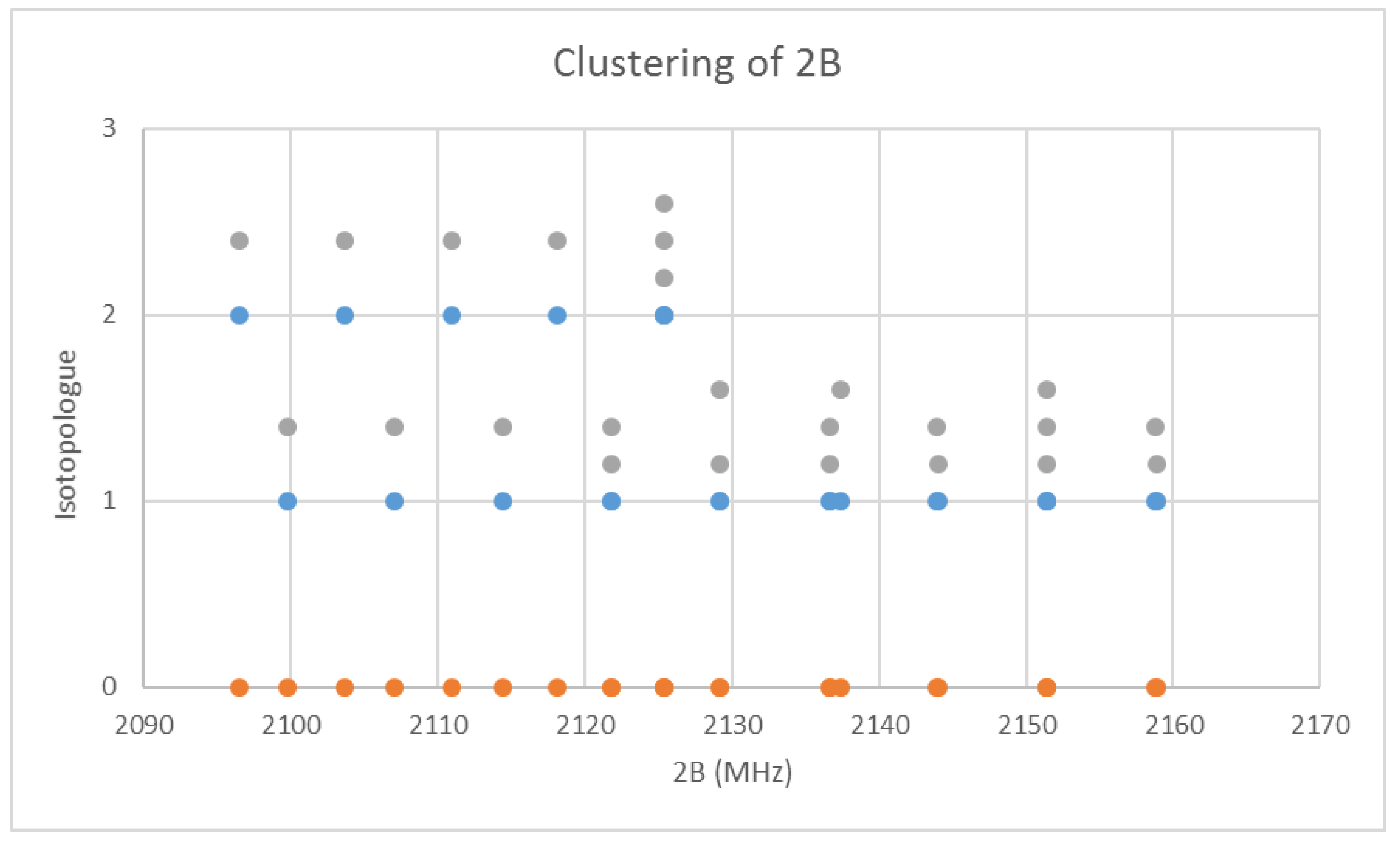
Bromine has two isotopes in approximately a 1:1 ratio, so two isotopologues should be observable in natural samples. The predicted value of 2B from the electron diffraction value is 2100 MHz (see Supplementary Material, Excel file CsBr.xlsx). The spectrum of cesium bromide in the region 21–26 GHz was measured [31] at 690 °C, and the frequencies observed (error 0.1–0.2 MHz) are given in Figure 4. Inspection of the 24 frequencies demonstrates that there are three clusters of frequencies centered at 21,382, 23,334, and 25,630 MHz [32]. The average difference of these is 2124 ± 150 MHz. The highest frequency per cluster differences is 2113 ± 45 MHz. These all suggest transitions corresponding to J = 9, 10, and 11.
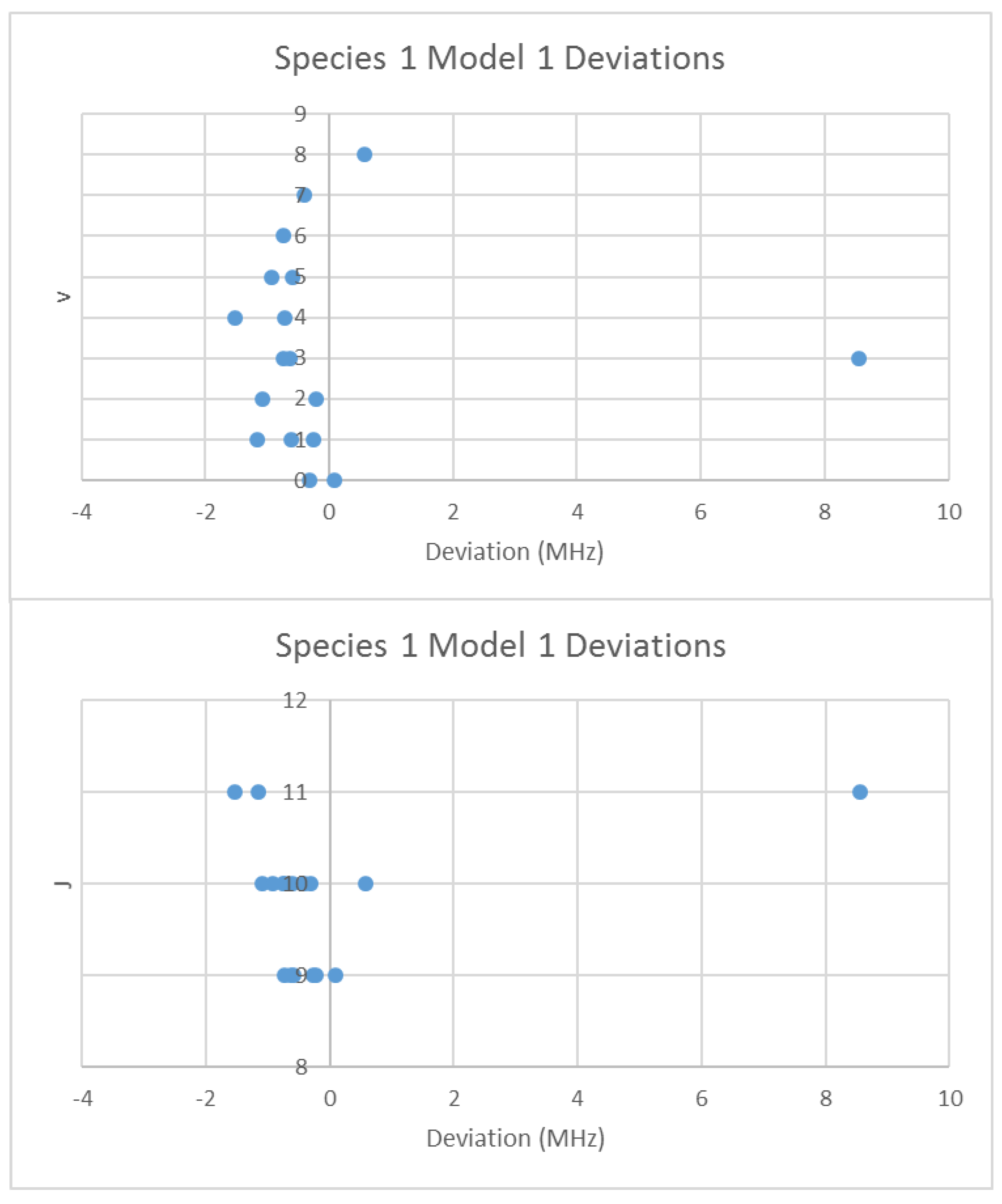
When the effective rotational constants (×2) are plotted (Figure 5), it can be seen that they naturally divide themselves into two series of equally spaced progressions, with the exception of a point at 2137.4 MHz corresponding to the observed frequency at 25,648.95 MHz. Fitting of these to Equation (37) gives Be values of 1081.27 and 1064.51 MHz. This was performed by fitting the data to both isotopologues and choosing the smaller of the two sum-of-squared errors. The deviation in 2B for the “wrong” isotopologue is an approximately constant 33 MHz. This procedure also enables separation by isotopologue. The isotopologue mass ratio is 1.015736, and the Be ratio is 1.015744, an agreement to five significant figures. This is strong evidence that the higher frequency progression (Isotopologue 1) is 133Cs79Br, and the lower frequency progression (Isotopologue 2) is 133Cs81Br. We can analyze these progressions separately. Using Model 1, the residual error for 133Cs79Br is plotted in Figure 6. The “bad” point corresponds to v = 3, J = 11. While a Q-test on the residuals can be used to exclude this point, it is important to note that this point appears to be 10 MHz too high. We suspect that this is a printing error, and that this value should actually be 25,638.95 MHz instead of 25,648.95 MHz. We will use this value henceforth as it reduces the SSE 20-fold. The results of fitting to all four models are given in Table 3. In all cases, the Be ratio matches the expected isotopic mass ratio by five significant figures. For Model 4, the re values for the two isotopologues, 3.072274 and 3.072287 Å, are in excellent agreement.
The pedagogical goals achieved here show that for CsBr, calculating effective rotational constants using Model 1 can (1) help assign points to different isotopologues, (2) identify a “bad” point, and (3) confirm isotopologue assignments by a calculation of Be ratios.
6.3. Cesium Chloride (CsCl)
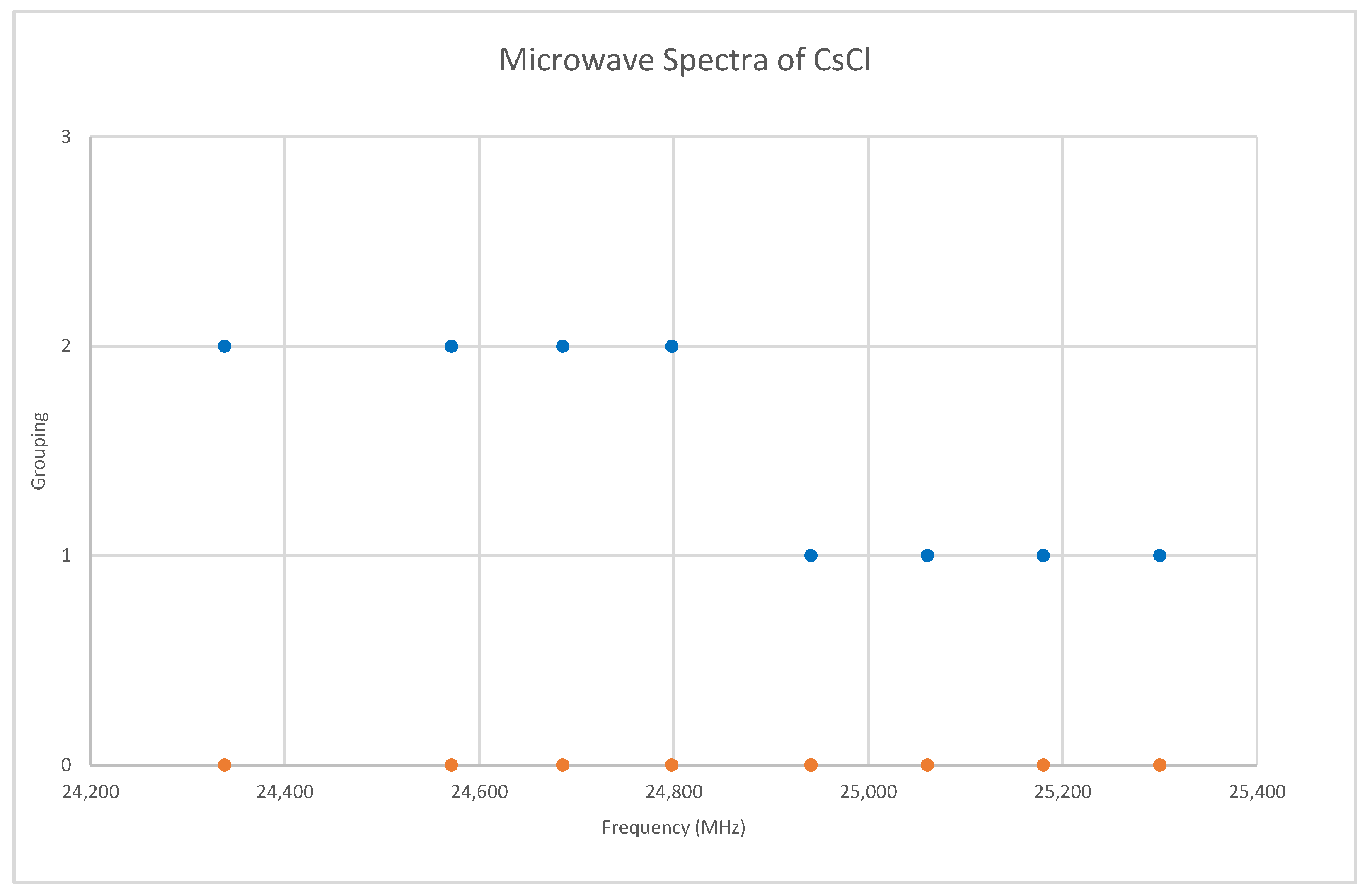
Chlorine has two isotopes in approximately a 3:1 ratio, so two isotopologues should be observable in natural samples. The predicted value of 2B from the electron diffraction value is 4000 MHz (See Supplementary Materials, Excel file CsCl.xlsx). The spectrum of cesium chloride in the region 24.3–25.4 GHz was measured [38] at 715 °C, and the frequencies observed (error 1.5 MHz) are given in Figure 7. Inspection of the eight frequencies demonstrates that there appears to be just one major grouping of frequencies. The estimated value of 2B suggests transitions corresponding to J = 5. There are two apparent gaps at about 24,450 and 24,880 MHz, which appear to be due to a missing transition and a break between two vibrational progressions, respectively. We note that because we do not have multiple values of J, there is no way to estimate the centrifugal distortion constant. We also note that the error in each measurement is fairly large compared to the previous two cesium salts analyzed.
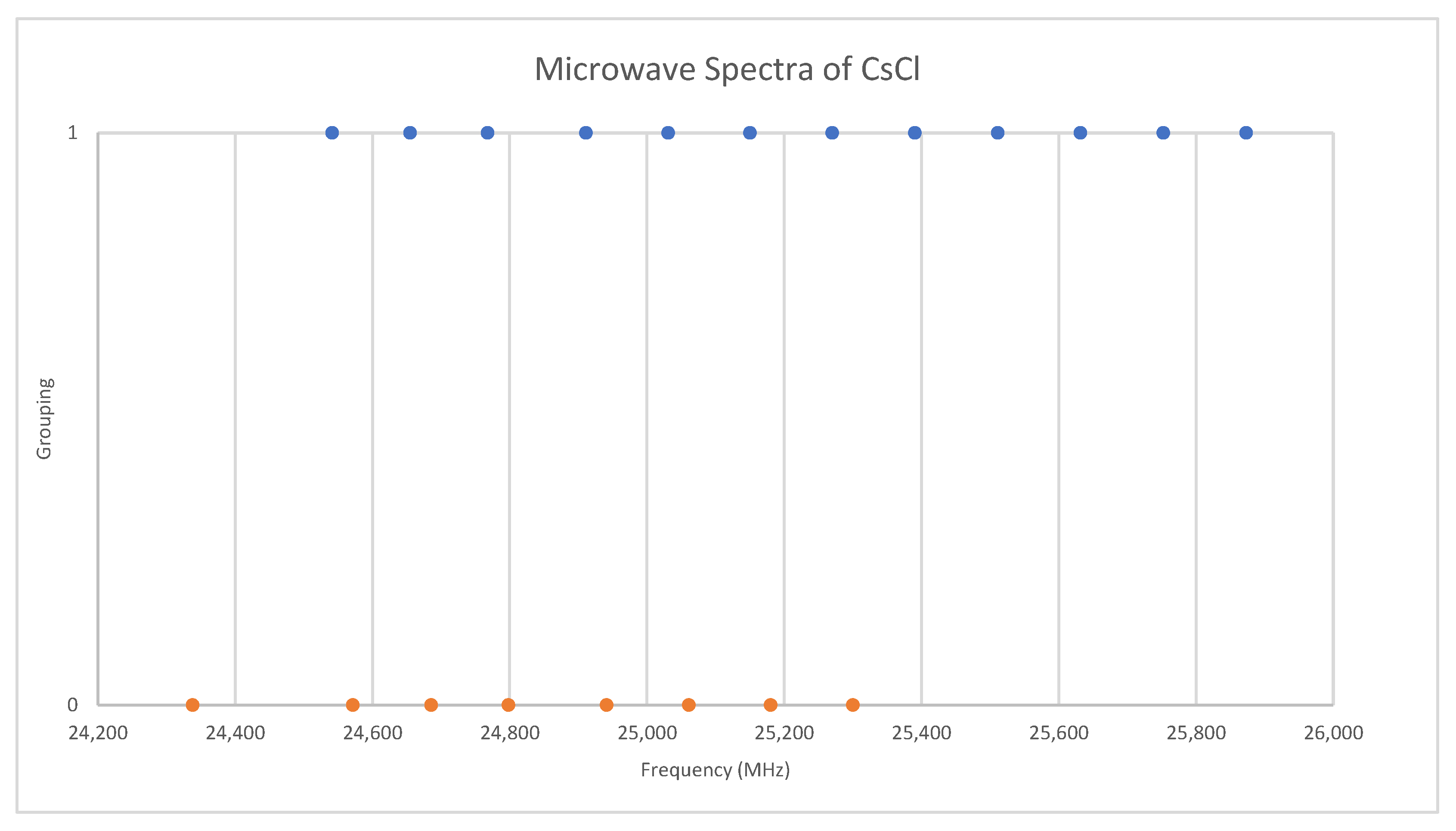
Under the assumption that the head of each progression corresponds to v = 0, we find that the higher frequency progression is well described by Model 1, with Be = 2113.31 and αe = 9.96, whereas the lower frequency progression gives Be = 2071.45 and αe = 9.60 (all MHz). The expected mass ratio is 1.04468, but the Be ratio is 1.02020. Therefore, the vibrational assignments are incorrect, and we must revise our assumption. It is unlikely that we would miss the head of the progression of the heavier isotopologue, as it falls within the range of other observed bands, so we assume that we have assigned this correctly and that we have misassigned the lighter isotopologue because it falls out of the observed range. With this model, any misassignment of v results in the same αe, but different Be. The following Be ratios are calculated for head assignments v = 1–5: 1.025, 1.029, 1.034, 1.039, and 1.044. This suggests that the assignments of the lighter isotopologue correspond to v = 5–8 instead of v = 0–3, and that the isotopologues are 133Cs35Cl and 133Cs37Cl. This was confirmed by a later measurement at 720 °C [31], where the missing transitions (v = 0–4) were seen. The newer results were all systematically lower in frequency by 30 MHz, but the precision was improved to 0.1–0.5 MHz (Figure 8). The model parameters are given in Table 4. For Model 2, the re values for the two isotopologues, 2.906337 and 2.906362 Å, are in excellent agreement.
The pedagogical goal achieved here is showing that the vibrational assignments should always be checked by comparison of the predicted Be ratios.
6.4. Cesium Fluoride (CsF)
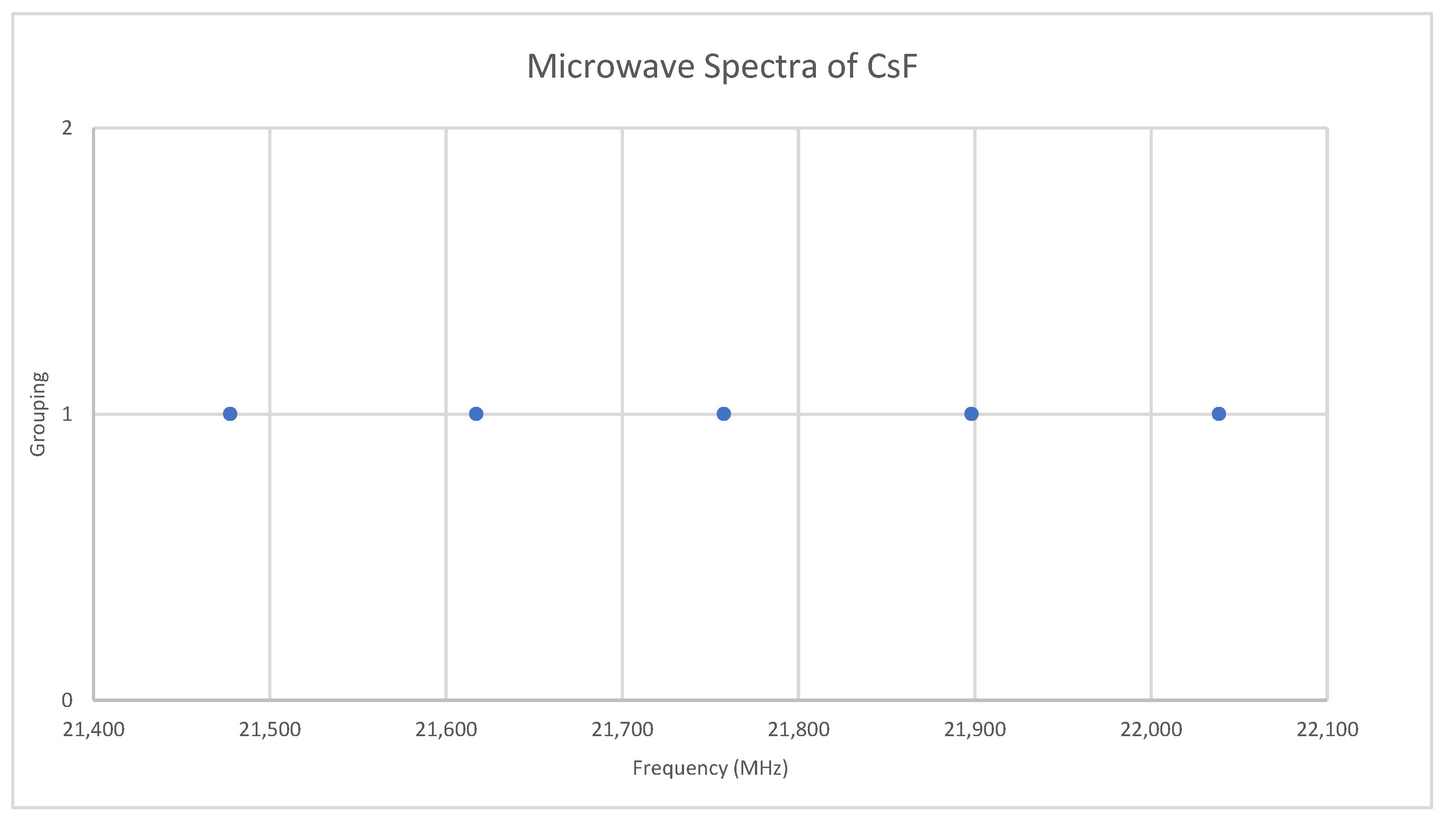
Fluorine is monoisotopic. The predicted value of 2B from the electron diffraction value is 10,000 MHz (See Supplementary Materials, Excel file CsF.xlsx). The spectra of cesium fluoride in the region 21.4–22.1 GHz was measured [31] at 700 °C, and the frequencies observed (error 0.2–1.0 MHz) are given in Figure 9. Inspection of the five frequencies demonstrates that there appears to be just one major grouping of frequencies. The observed spectrum corresponds to J = 1, v = 0–4. Fitting gives Be = 5527.123, αe = 35.072 (Model 1), and Be = 5527.259, αe = 35.218, γe = 0.0279 (Model 2), all MHz. The bond distance re is 2.3454 Å. This problem makes an excellent student exercise (Appendix A) without the complicating effects of multiple J and/or isotopes.
6.5. Rubidium Iodide (RbI)
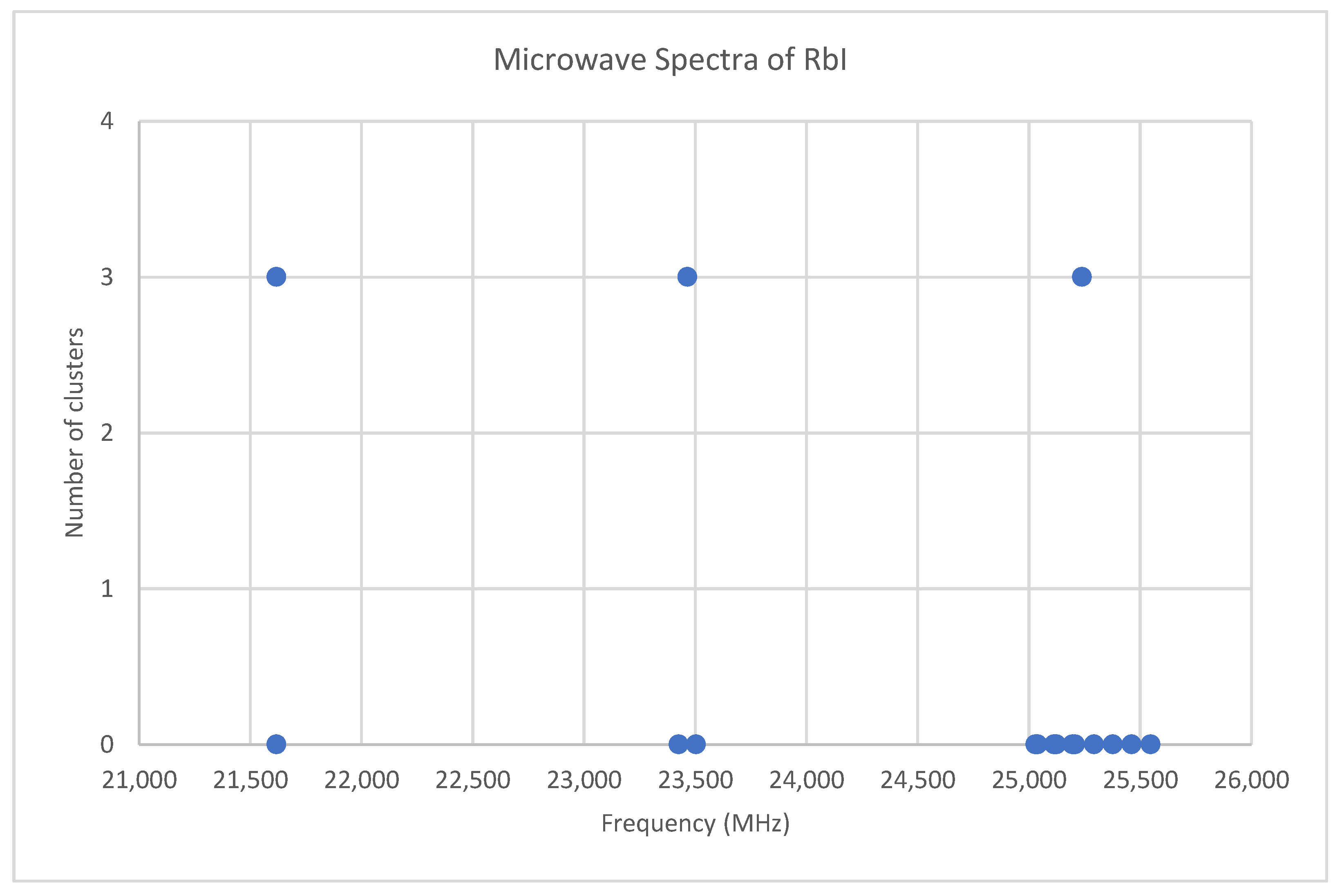
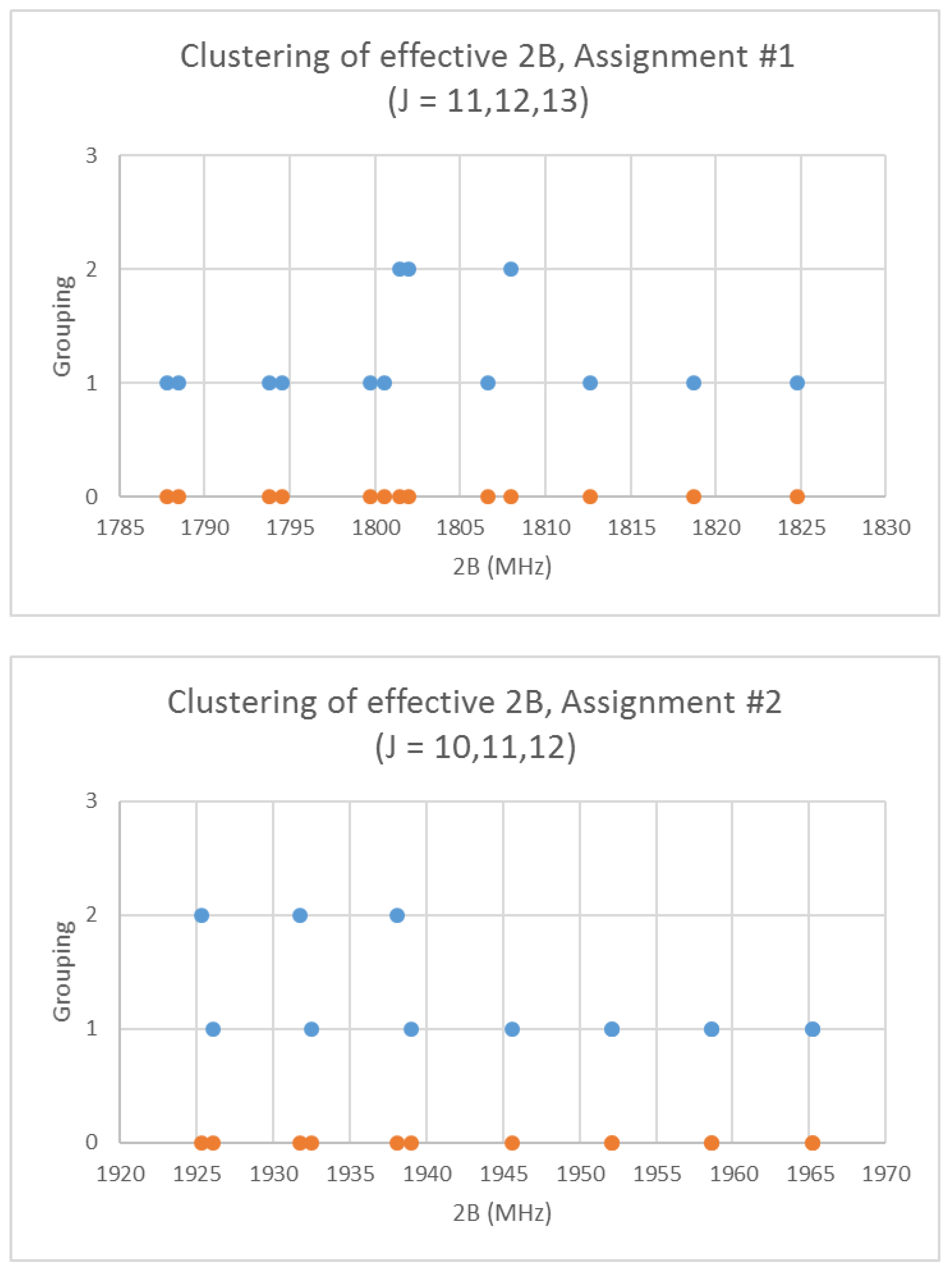
Rubidium has two isotopes, 85Rb and 87Rb, present in approximately a 3:1 ratio. The predicted value of 2B from electron diffraction is 1880 MHz (see Supplementary Materials, Excel file RbI.xlsx). The spectra of rubidium iodide in the region 21.5–25.6 GHz was measured [31] at 660 °C, and the frequencies observed (error 0.1–0.2 MHz) are given in Figure 10. Inspection of the 13 frequencies demonstrates that there appears to be three major groupings (clusters) of frequencies. The centers of the clusters are at 21,617.58, 23,464.75, and 25,238.83 MHz, with an average difference of 1810(37) MHz. The average difference between the highest frequency components is 1965(80) MHz. Using the estimated 2B or cluster averages would suggest either J = 10, 11, 12 (Assignment # 2) or J = 11, 12, 13 (Assignment #1), whereas using the highest frequency differences (assuming v = 0) would suggest J = 10, 11, 12, with an anomaly for J = 11. While the preference is for J = 10, 11, 12, the assignment is somewhat less definitive, so we have plotted the effective rotational constants for both scenarios (Figure 11). For assignment #1, all 13 points are still visible in nine subclusters, apparently in two vibrational progressions. This would imply that the centrifugal distortion constants are quite large, and that there are at least four points with the same v, but with multiple values of J. In addition, the Be ratio is 1.009306. For assignment #2, only 10 clusters are visible, and these can (barely) be separated into two vibrational progressions with a Be ratio of 1.013951. The expected mass ratio is 1.013961, which agrees with the second assignment to five significant figures. We therefore assume that the transitions are as assignment #2. The effective rotational constants for J = 11 correspond to v = 1–2, not to v = 0–1, which explains why the highest frequency method for J determination did not work as well.
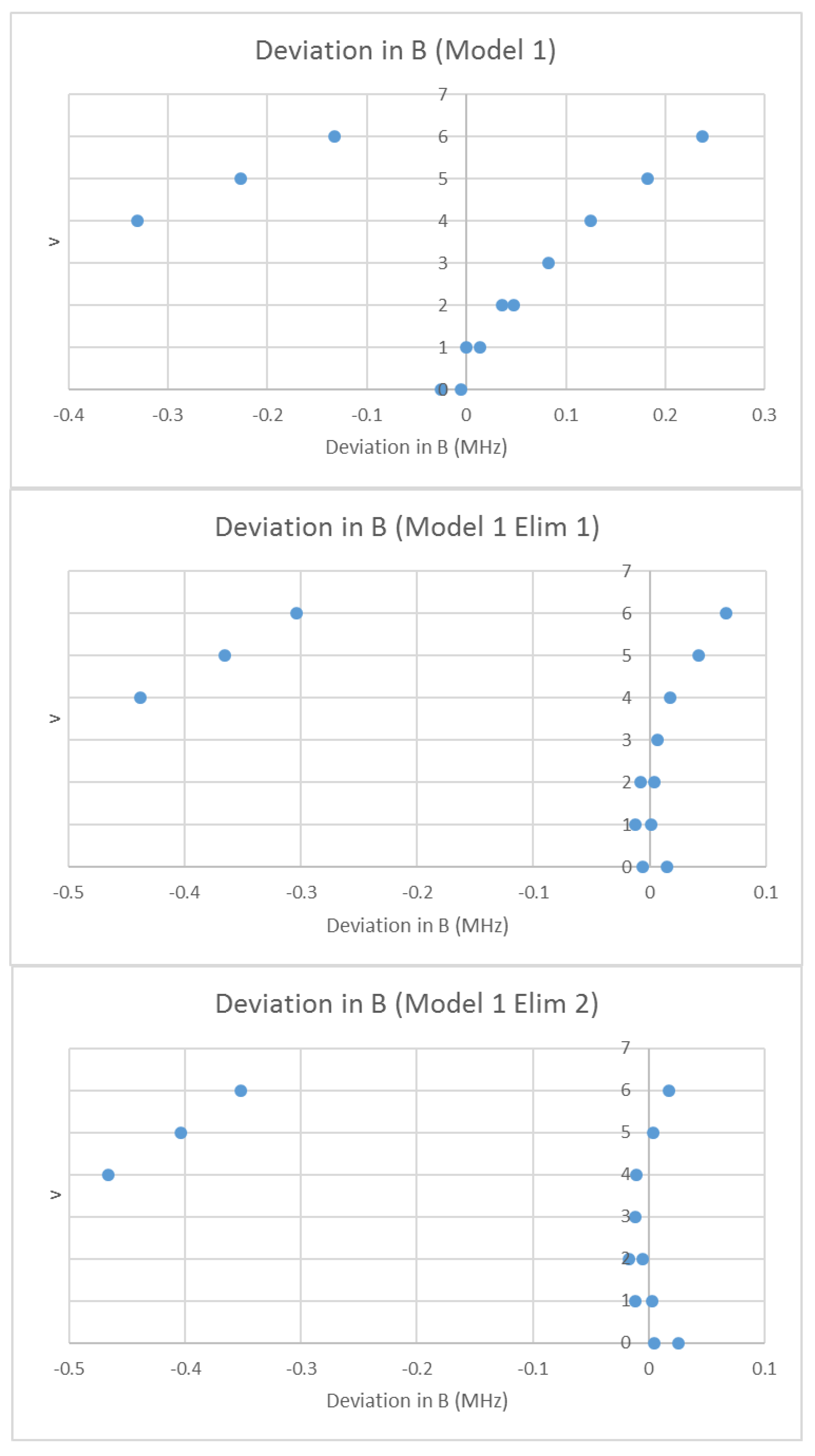
What would happen if we assumed that the spectrum was due to a single isotopologue, but that some lines were doubled due to some unknown effect? In that case, we may analyze the effective rotational constants as usual using Model 1. The residual error of this model is shown in Figure 12. There is a clear separation of the residual into two groups with this double assignment, and the least-squares process tries to fit both, resulting in a fairly large error. If we eliminate all points currently assigned to v = 4–6 (Model 1 Elim 1), the error drops significantly, and one set of the excluded points fits the expected residual error pattern. We can then re-include those points in the fit (Model 1 Elim 2). The points excluded belong to a second species. We then re-assign the vibrational quantum numbers of the second species to obtain a reasonable Be ratio, from v = 4–6 (1.000736) to v = 0–2 (1.013950). We then proceed as usual to obtain re of 3.176993 and 3.176997 Å for the two isotopologues (Table 5).
The pedagogical goals achieved here show that that, for RbI, (1) plotting effective rotational constants for different J assignments can assist in determining J, and (2) plotting residuals can help identify incorrect assumptions.
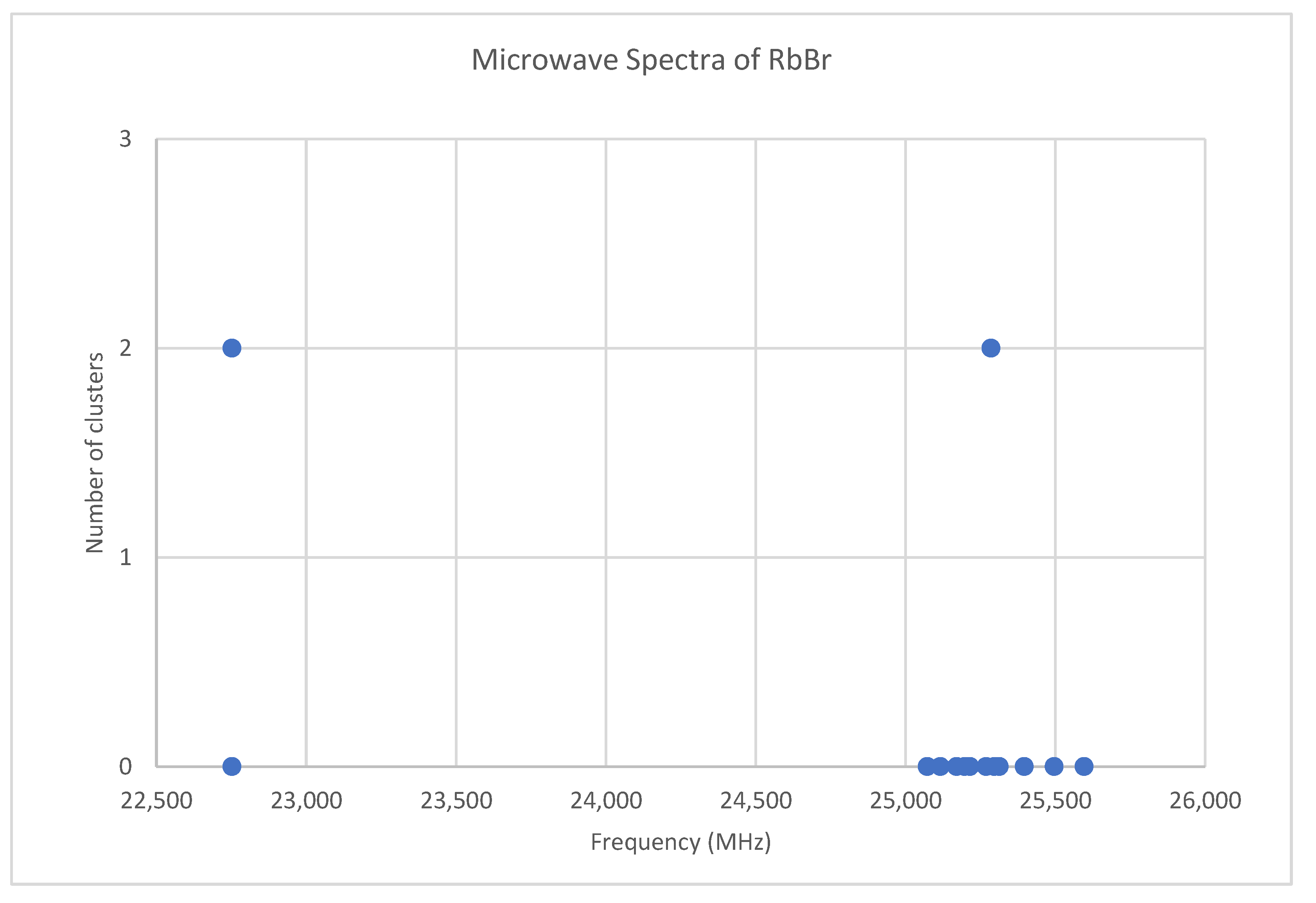
6.6. Rubidium Bromide (RbBr)
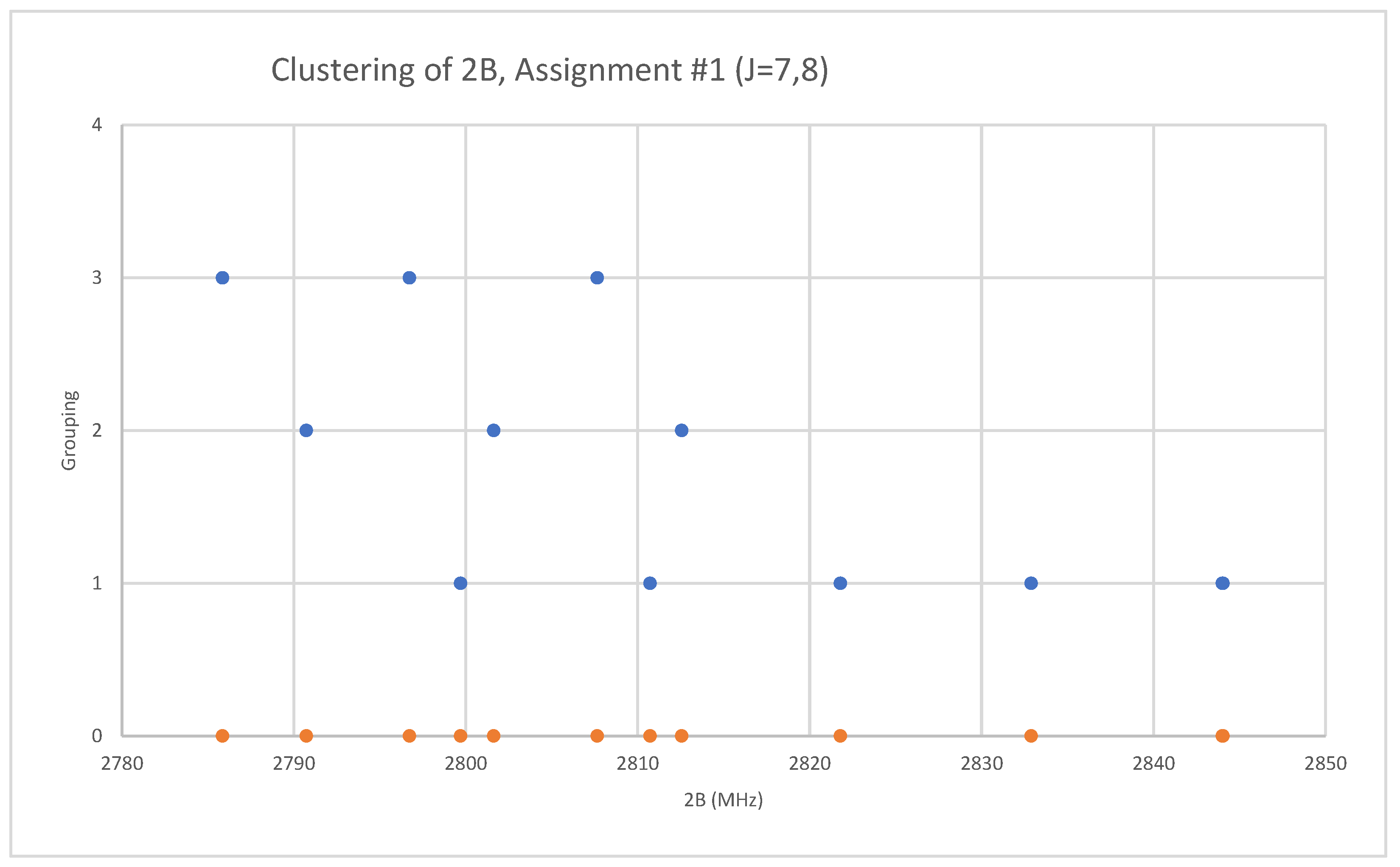
Because both rubidium and bromine are diisotopic, there may be a total of four isotopologues that could be observed. The predicted value of 2B from electron diffraction is 2650 MHz (see Supplementary Materials, Excel file RbBr.xlsx). The spectrum of rubidium bromide in the region 22.5–26.0 GHz was measured [31] at 730 °C, and the frequencies observed (error 0.1 MHz) are given in Figure 13. Inspection of the 12 frequencies demonstrates that there appear to be two major groupings (clusters) of frequencies, a single frequency at 22,752.29 MHz and a cluster centered at 25,285.29 MHz. The cluster difference is 2533 MHz, but the highest frequency components differ by 2843.70 MHz. Using the estimated 2B suggests either J = 7,8 or J = 8,9. Using the cluster average would suggest J = 8,9, whereas using the highest frequency differences (assuming v = 0) would suggest J = 7,8. With the highest frequency differences usually being more accurate, and along with the 2B clustering favoring this as well, we assign these to J = 7,8, with three different vibrational progressions (Figure 14).
The lightest isotopologue (85Rb79Br) is also the most abundant. The mass ratios for the heavier isotopologues are 1.011195 (87Rb79Br), 1.012963 (85Rb81Br), and 1.024452 (87Rb81Br). The Be ratios for Model 1 are calculated as 1.011188 and 1.012955 and for Model 2 as 1.011202 and 1.012965, which demonstrate that the other two isotopologues observed are 87Rb79Br and 85Rb81Br (Table 6). The values of re are calculated as 2.944776, 2.944820, and 2.944823 Å.
The pedagogical goal achieved here shows that, for RbBr, more than two isotopologues can be successfully analyzed.
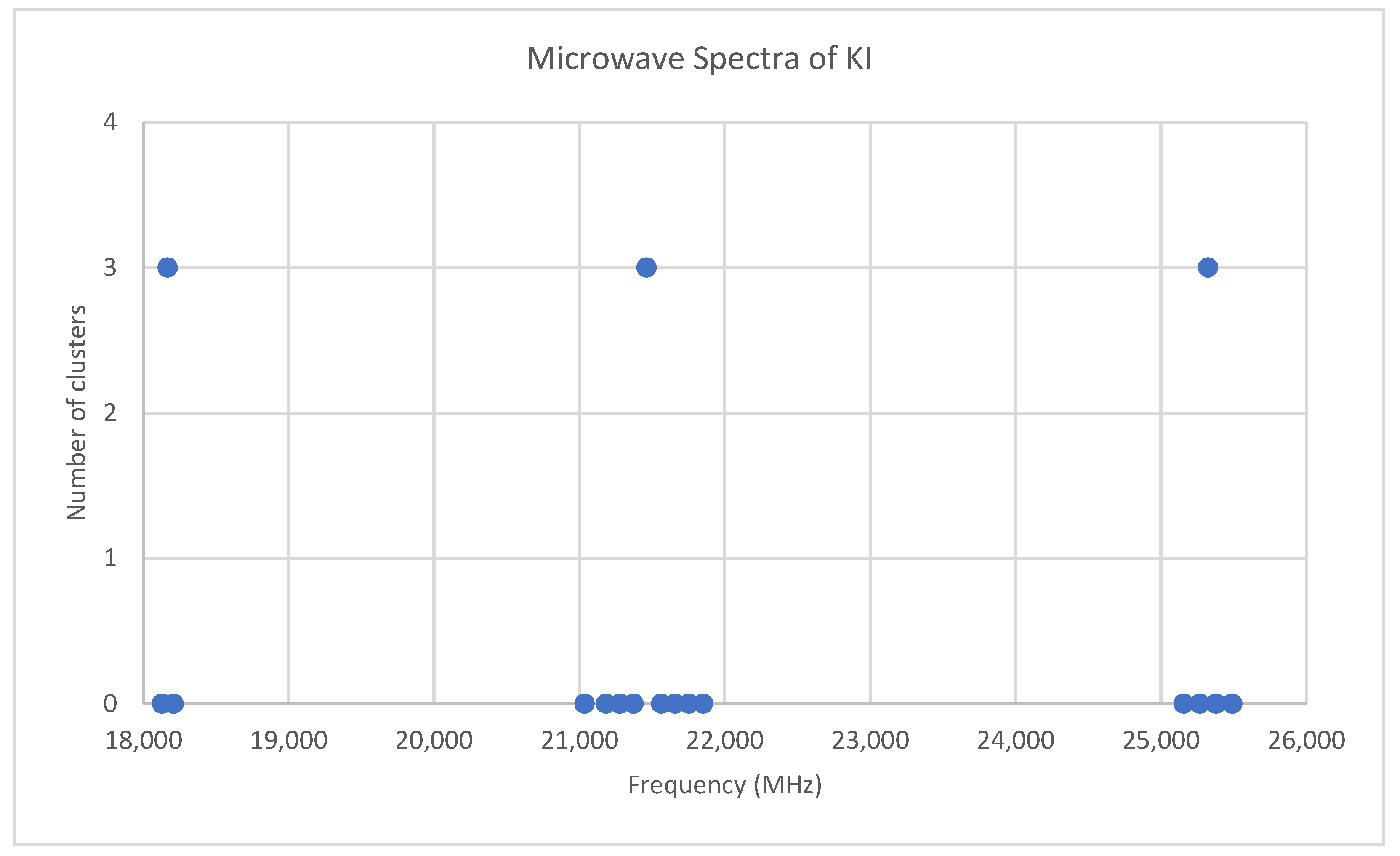
6.7. Potassium Iodide (KI)
Potassium has two isotopes, 39K and 41K, present in approximately a 14:1 ratio. The predicted value of 2B from electron diffraction is 3300 MHz (see Supplementary Materials, Excel file KI.xlsx). The spectrum of potassium iodide in the region 18–26 GHz was measured [31] at 690 °C, and the frequencies observed (error 0.1–0.3 MHz) are given in Figure 15. Inspection of the 14 frequencies demonstrates that there appears to be three major groupings (clusters) of frequencies. Analysis of this spectrum is left for the reader as a more challenging exercise (Appendix B). The results for 39K127I are Model 1, Be = 1824.840, αe = 7.941 MHz; Model 2, Be = 1824.962, αe = 8.047, γe = −0.0141 MHz; Model 3, Be = 1825.023, αe = 7.944, De = 0.0022 MHz; Model 4, Be = 1825.059, αe = 8.040, γe = −0.0130, De = 0.0013 MHz. The value of re is calculated as 3.047776 Å. The value of r0 for the heavier isotopologue 41K127I can be calculated as 3.051148 Å.
6.8. Potassium Chloride (KCl)
Potassium chloride has four naturally occurring isotopologues: 39K35Cl (70.65%), 39K37Cl (22.61%), 41K35Cl (5.10%), 41K37Cl (1.63%). The predicted value of 2B from electron diffraction is 7100 MHz (see Supplementary Materials, Excel file KCl.xlsx). The spectra of potassium chloride in the region 22.4–23.2 GHz was measured [39] at 715 °C, and the four frequencies observed (error 10 MHz) could either correspond to a vibrational progression, with a gap, or possibly to a mixture of up to four isotopologues. This determination is left to the reader (see Appendix C). A more precise spectrum was measured [40] at 700 °C and is also left as an exercise (see Appendix D).
6.9. Sodium Chloride (NaCl)
Sodium is monoisotopic. The predicted value of 2B from electron diffraction is 11,600 MHz (see Supplementary Materials, Excel file NaCl.xlsx). The spectrum of sodium chloride in the region 25.0–26.1 GHz was measured [38] at 775 °C, and the seven frequencies observed (error 0.75 MHz) correspond to two vibrational progressions. The assignment is left to the reader (Appendix E).
7. Conclusions
The microwave spectra of CO, CsI, CsBr, CsCl, CsF, RbI, RbBr, KI, KCl, and NaCl are presented and procedures for their analysis are discussed. By the use of either additional information (electron or X-ray diffraction) or by cluster analysis of the spectra, the approximate value of 2B can be determined, followed by the assignment of the frequencies to rotational quantum number J (Model 0). Once assigned, effective rotational constants for each transition can be assigned to specific vibrational quantum numbers v and/or isotopologues, to obtain the rotational constant Be, bond length re, and vibration–rotation interaction constant αe (Model 1). In some cases, other spectroscopic constants (De, γe) can be determined.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/spectroscj1010002/s1, Microsoft Excel spreadsheets CO.xlsx, CsI.xlsx, CsBr.xlsx, CsCl.xlsx, CsF.xlsx, RbI.xlsx, RbBr.xlsx, KI.xlsx, KCl.xlsx, NaCl.xlsx, Isotopes.xlsx.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data used in this article can be found in either the references cited or in the Supplementary Material.
Acknowledgments
CCP acknowledges Danielle Tokarz, Department of Chemistry, Saint Mary’s University, for suggesting corrections to this manuscript prior to submission.
Conflicts of Interest
The author declares no conflict of interest.
Appendix A. Analysis of the Microwave Spectrum of Cesium Fluoride (CsF)
The microwave spectrum of cesium fluoride in the range 21.4–23.0 GHz has transitions at 22,038.51, 21,898.21, 21,757.58, 21,617.09, and 21,477.5 MHz. X-ray diffraction of cesium fluoride gives a Cs-F distance of 3.004 Å. X-ray diffraction typically gives M-X distances that are 15–20% longer than gas-phase electron diffraction for the alkali halides. (a) Predict the value of 2B that should be observed. (b) Assign the observed transitions to definite rotational and vibrational quantum numbers. (c) Calculate Be and αe. (d) Give a precise value of re.
[Ans: (a) 10 GHz (b) J = 1, v = 0–4 (c) Model 2 (MHz): Be = 5527.259, αe = 35.218 (d) re = 2.3454 Å]
Appendix B. Analysis of the Microwave Spectrum of Potassium Iodide (KI)
The microwave spectrum of potassium iodide in the range 18–26 GHz has transitions at 18,129.61, 18,209.77, 21,036.78, 21,184.73, 21,279.07, 21,373.63, 21,563.91, 21,659.38, 21,755.19, 21,851.32, 25,157.04, 25,268.95, 25,380.71, and 25,492.81 MHz. Electron diffraction gives a K-I distance of 3.23 Å. (a) Predict the value of 2B that should be observed. (b) Assign the transitions to definite isotopologues, rotational and vibrational quantum numbers. (c) Calculate Be, αe, γe, De where possible. (d) Give a precise value of re.
[Ans: (a) 3.3 GHz (b) All due to 39K127I (J = 4, v = 0,1; J = 5, v = 0–3, 5–7; J = 6, v = 0–3) except 21,036.78 MHz (41K127I, J = 5, v = 0) (c) 39K127I Model 4 (MHz): Be = 1825.059, αe = 8.040, γe = −0.013, De = 0.00129 (d) re = 3.047776 Å]
Appendix C. Analysis of the Microwave Spectrum of Potassium Chloride (KCl)
A low-resolution (10 MHz) microwave spectrum of potassium chloride gives transitions at 23,066, 22,918, 22,646, and 22,504 MHz, corresponding to J = 2→3. By calculating spectroscopic parameters and isotopologue mass ratios, determine whether these transitions are more likely due to a vibrational progression of a single isotopologue or to a mixture of isotopologues.
[Ans: vibrational progression of a single isotopologue with a missing point J = 2, v = 0,1,3,4; Model 2 (MHz): Be = 3858.1, αe = 27.2, γe = 0.7]
Appendix D. Analysis of the Microwave Spectrum of Potassium Chloride (KCl)
A medium resolution (<3 MHz) microwave spectrum of potassium chloride gives transitions at 22,278.0, 22,410.3, 22,644.0, 22,785.2, 22,925.4, and 23,067.5 MHz, corresponding to J = 2→3. By calculating spectroscopic parameters and isotopologue mass ratios, determine whether these transitions are more likely due to a vibrational progression of a single isotopologue or to a mixture of isotopologues.
[Ans: mixture of two isotopologues 39K35Cl (head 23,067.5 MHz, v = 0–3) and 39K37Cl (head 22,410.3, v = 0,1), Model 1 (MHz): 39K35Cl, Be = 3856.278, αe = 23.512; 39K37Cl, Be = 3746.075, αe = 22.050 (d) re = 2.6667Å]
Appendix E. Analysis of the Microwave Spectrum of Sodium Chloride (NaCl)
A medium-resolution (0.75 MHz) microwave spectrum of sodium chloride gives transitions at 25,120.3, 25,307.5, 25,473.9, 25,493.9, 25,666.5, 25,857.6, and 26,051.1 MHz, corresponding to J = 1→2. How many isotopologues do you expect to observe? Assign the spectra and calculate the appropriate molecular constants.
[Ans: Two, 23Na35Cl, head 26,051.1 MHz, v = 0–4 and 23Na37Cl, head 25,493.9 MHz, v = 0–2. Model 1 (MHz): 23Na35Cl, Be = 6536.70, αe = 48.07; 23Na37Cl, Be = 6396.86, αe = 46.7 (d) re = 2.3609 Å]
References
- Ewing, G.W. Microwave Absorption Spectroscopy. J. Chem. Educ. 1966, 43, A683–A722. [Google Scholar]
- Townes, C.H.; Schawlow, A.L. Microwave Spectroscopy; Dover: New York, NY, USA, 1975. [Google Scholar]
- Schwendeman, R.H.; Volltrauer, H.N.; Laurie, V.W.; Thomas, E.C. Microwave Spectroscopy in the Undergraduate Laboratory. J. Chem. Educ. 1970, 47, 526–532. [Google Scholar]
- Dyke, T.R.; Muenter, J.S. A Stark Modulation Absorption Cell for Student Microwave Spectrometers. J. Chem. Educ. 1974, 51, 33. [Google Scholar] [CrossRef]
- Pollnow, G.F.; Hopfinger, A.J. A Computer Experiment in Microwave Spectroscopy. J. Chem. Educ. 1968, 45, 528–531. [Google Scholar] [CrossRef]
- Pollnow, G.F.; Chung, C.S.C. An Interactive Computer Program for Microwave Spectroscopy. J. Chem. Educ. 1973, 50, 794. [Google Scholar]
- McNaught, I.J.; Moore, R. Microwave Spectroscopy Tutor. J. Chem. Educ. 1995, 72, 993–994. [Google Scholar] [CrossRef]
- McNaught, I.J.; Moore, R. Winspec: A Microwave Spectroscopy Tutor. J. Chem. Educ. 1996, 73, 523.az. [Google Scholar]
- Woods, R.; Henderson, G. FTIR Rotational Spectroscopy. J. Chem. Educ. 1987, 64, 921–924. [Google Scholar] [CrossRef]
- McQuarrie, D.A. Quantum Chemistry, 2nd ed.; University Science Books: Herndon, VA, USA, 2008; Chapter 6. [Google Scholar]
- Pye, C.C. On the Solution of the Quantum Rigid Rotor. J. Chem. Educ. 2006, 83, 460–463. [Google Scholar] [CrossRef]
- Moynihan, C.T. Rationalization of the ΔJ=±1 Selection Rule for Rotational Transitions. J. Chem. Educ. 1969, 46, 431. [Google Scholar]
- Foss, J.G. Photonic Angular Momentum and Selection Rules for Rotational Transitions. J. Chem. Educ. 1970, 47, 778–779. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Sannigrahi, A.B. A Simple Group-Theoretical Derivation of the Selection Rules for Rotational Transitions. J. Chem. Educ. 1990, 67, 653–655. [Google Scholar]
- Laidler, K.J.; Meiser, J.H. Physical Chemistry, 3rd ed.; Houghton Mifflin: Boston, MA, USA, 1999; Chapter 13. [Google Scholar]
- Engel, T.; Reid, P. Physical Chemistry; Pearson: San Francisco, CA, USA, 2006; Chapter 19. [Google Scholar]
- House, J.E. Fundamentals of Quantum Mechanics; Academic Press: San Diego, CA, USA, 1998; Chapter 7. [Google Scholar]
- Ratner, M.A.; Schatz, G.C. Introduction to Quantum Mechanics in Chemistry; Prentice-Hall: Upper Saddle River, NJ, USA, 2001; Chapter 4. [Google Scholar]
- Atkins, P.; de Paula, J. Physical Chemistry, 10th ed.; Freeman: New York, NY, USA, 2014; Chapter 12. [Google Scholar]
- Berry, R.S.; Rice, S.A.; Ross, J. Physical Chemistry, 2nd ed.; Oxford University Press: New York, NY, USA, 2000; Chapter 7. [Google Scholar]
- Alberty, R.A.; Silbey, R.J. Physical Chemistry, 2nd ed.; Wiley: New York, NY, USA, 1997; Chapter 13. [Google Scholar]
- Mortimer, R.G. Physical Chemistry, 2nd ed.; Harcourt: San Diego, CA, USA, 2000; Chapter 19. [Google Scholar]
- Zielinski, T.J. Fostering Creativity and Learning Using Instructional Symbolic Mathematics Documents. J. Chem. Educ. 2009, 86, 1466–1467. [Google Scholar]
- Microsoft Office Professional Plus (Excel) 2016, Microsoft Corporation: Redmond, WA, USA, 2016.
- Solver, Microsoft Office Add-In, Frontline Systems: Incline Village, NV, USA, 2016.
- Gilliam, O.R.; Johnson, C.M.; Gordy, W. Microwave Spectroscopy in the Region from Two to Three Millimeters. Phys. Rev. 1950, 78, 140–144. [Google Scholar]
- Vegard, L. Struktur und Leuchtfahigkeit von festem Kohlenoxyd. Z. Phys. 1930, 61, 185–190. [Google Scholar] [CrossRef]
- Crystallography Open Database. Available online: http://www.crystallography.net (accessed on 31 March 2018).
- Wyckoff, R.W.G. Crystal Structures; Wiley: New York, NY, USA, 1963; Chapter 3; pp. 85–237. [Google Scholar]
- Maxwell, L.R.; Hendricks, S.B.; Mosley, V.M. Interatomic Distances of the Alkali Halide Molecules by Electron Diffraction. Phys. Rev. 1937, 52, 968–972. [Google Scholar]
- Honig, A.; Mandel, M.; Stitch, M.L.; Townes, C.H. Microwave Spectra of the Alkali Halides. Phys. Rev. 1954, 96, 629–642. [Google Scholar] [CrossRef]
- Pye, C.C. One-Dimensional Cluster Analysis and its Application to Chemistry. Chem. Educ. 2020, 25, 50–57. [Google Scholar]
- Pye, C.C. Intuitive Solution to Quantum Harmonic Oscillator at Infinity. J. Chem. Educ. 2004, 81, 830–831. [Google Scholar]
- McHale, J.L. Molecular Spectroscopy; Prentice-Hall: Upper Saddle River, NJ, USA, 1999; Chapters 8–9. [Google Scholar]
- Dykstra, C.E. Introduction to Quantum Chemistry; Prentice-Hall: Englewood Cliffs, NJ, USA, 1994; Chapter 5. [Google Scholar]
- Hollenberg, J.L. Energy States of Molecules. J. Chem. Educ. 1970, 47, 2–14. [Google Scholar] [CrossRef]
- Brown, J.M. Molecular Spectroscopy; Oxford Science Publications: Oxford, UK, 1999; Chapter 5. [Google Scholar]
- Stitch, M.L.; Honig, A.; Townes, C.H. Microwave Spectra at High Temperature—Spectra of CsCl and NaCl. Phys. Rev. 1952, 86, 813–814. [Google Scholar] [CrossRef]
- Stitch, M.L.; Honig, A.; Townes, C.H. High Temperature Microwave Spectroscopy—Spectrum of KCl, TlCl. Phys. Rev. 1952, 86, 607. [Google Scholar]
- Tate, P.A.; Strandberg, M.W.P. Stark Effect in the Microwave Spectra of KCl and NaCl. J. Chem. Phys. 1954, 22, 1380–1383. [Google Scholar] [CrossRef]
Figure 1.
The spectrum of cesium iodide (Grouping 0), and the cluster averages for k = 3.

Figure 2.
The effective rotational constants (×2) of cesium iodide.

Figure 3.
The residual plots of four different models for the microwave spectrum of cesium iodide.

Figure 4.
The microwave spectrum of cesium bromide, and the 3-cluster averages.

Figure 5.
The effective rotational constants (×2) of cesium bromide. Different J values are offset by 0.2 units. The orange points are the original spectrum, the blue points are separation into isotopologues.
Figure 5.
The effective rotational constants (×2) of cesium bromide. Different J values are offset by 0.2 units. The orange points are the original spectrum, the blue points are separation into isotopologues.

Figure 6.
The deviation from Model 1 for 133Cs79Br.

Figure 7.
The microwave spectrum of cesium chloride, and separation into isotopologues. The orange points are the original spectrum, the blue points are separation into isotopologues.
Figure 7.
The microwave spectrum of cesium chloride, and separation into isotopologues. The orange points are the original spectrum, the blue points are separation into isotopologues.

Figure 8.
Comparison of microwave spectra of cesium chloride from two sources (Grouping 0 (orange) [38], Grouping 1 (blue) [31]).

Figure 9.
The microwave spectrum of cesium fluoride.

Figure 10.
The microwave spectrum of rubidium iodide, and the 3-cluster averages.

Figure 11.
The rotational constants of rubidium iodide for two different J assignments. The orange points are the original spectrum, the blue points are separation into isotopologues.
Figure 11.
The rotational constants of rubidium iodide for two different J assignments. The orange points are the original spectrum, the blue points are separation into isotopologues.

Figure 12.
The Model 1 residuals of the effective rotational constants of rubidium iodide, assuming a single species. The top graph includes all points, the middle graph eliminates points assigned as v = 4–6 from the fit, and the bottom graph only eliminates the leftmost v = 4–6 points.
Figure 12.
The Model 1 residuals of the effective rotational constants of rubidium iodide, assuming a single species. The top graph includes all points, the middle graph eliminates points assigned as v = 4–6 from the fit, and the bottom graph only eliminates the leftmost v = 4–6 points.

Figure 13.
The microwave spectrum of rubidium bromide, and the 2-cluster averages.

Figure 14.
The rotational constants of rubidium bromide. The orange points are the original spectrum, the blue points are separation into isotopologues.
Figure 14.
The rotational constants of rubidium bromide. The orange points are the original spectrum, the blue points are separation into isotopologues.

Figure 15.
The microwave spectrum of potassium iodide, and the 3-cluster averages.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
The MX distance in alkali halides (M = Li, Na, K, Rb, Cs; X = F, Cl, Br, I).
| MX | R (Å) 1 | R (Å) 4 | Ratio |
|---|---|---|---|
| LiF | 2.0086 | ||
| LiCl | 2.56477 | ||
| LiBr | 2.7506 | ||
| LiI | 3.000 | ||
| NaF | 2.310 | ||
| NaCl | 2.8203 | 2.51 | 1.124 |
| NaBr | 2.98662 | 2.64 | 1.131 |
| NaI | 3.2364 | 2.90 | 1.116 |
| KF | 2.6735 | ||
| KCl | 3.14647 | 2.79 | 1.128 |
| KBr | 3.3000 | 2.94 | 1.122 |
| KI | 3.53278 | 3.23 | 1.093 |
| RbF | 2.82 | ||
| RbCl | 3.2905 | 2.89 | 1.139 |
| RbBr | 3.427 | 3.06 | 1.120 |
| RbI | 3.671 | 3.26 | 1.126 |
| CsF | 3.004 | ||
| CsCl | 3.51 2 | 3.06 | 1.147 |
| 3.5706 3 | 1.167 | ||
| CsBr | 3.7118 3 | 3.14 | 1.182 |
| CsI | 3.9549 3 | 3.41 | 1.159 |
1 X-ray diffraction (NaCl-like). 2 450 °C. 3 X-ray diffraction (CsCl-like). 4 Electron diffraction (gas), 1000–1300 °C.
Table 2.
Model parameters for cesium iodide (MHz) (133Cs127I, n = 9).
| Model | Be | αe | γe | De1 | SSE |
|---|---|---|---|---|---|
| 1 | 708.266 | 2.040 | n/a | n/a | 0.7213 |
| 2 | 708.269 | 2.045 | 0.0015 | n/a | 0.6840 |
| 3 | 708.280 | 2.040 | n/a | 25.7 | 0.5194 |
| 4 | 708.362 | 2.043 | 0.0011 | 162 | 0.0102 |
1 Centrifugal distortion constants have been multiplied by 106.
Table 3.
Model parameters for cesium bromide (MHz) (133Cs79Br, n = 17; 133Cs81Br, n = 7).
| Model | Be | αe | γe | De1 | SSE | |
|---|---|---|---|---|---|---|
| 133Cs79Br | 1 | 1081.242 | 3.692 | n/a | n/a | 3.998 |
| 2 | 1081.283 | 3.720 | 0.0033 | n/a | 1.426 | |
| 3 | 1081.281 | 3.692 | n/a | −0.162 | 2.520 | |
| 4 | 1081.313 | 3.718 | 0.0030 | −0.138 | 0.361 | |
| 133Cs81Br | 1 | 1064.512 | 3.617 | n/a | n/a | 0.263 |
| 2 | 1064.521 | 3.632 | 0.0032 | n/a | 0.177 | |
| 3 | 1064.538 | 3.618 | n/a | −0.097 | 0.130 | |
| 4 | 1064.552 | 3.635 | 0.0037 | −0.107 | 0.018 |
1 Centrifugal distortion constants have been multiplied by 106.
Table 4.
Model parameters for cesium chloride (MHz).
| Model | Be | αe | γe | SSE | |
|---|---|---|---|---|---|
| 133Cs35Cl [38], n = 4 | 1 | 2163.119 | 9.963 | n/a | 0.115 |
| [31], n = 9 | 1 | 2161.029 | 10.021 | n/a | 4.195 |
| 2 | 2161.152 | 10.10 | 0.0091 | 0.522 | |
| 133Cs37Cl [38], n = 4 | 1 | 2071.458 | 9.600 | n/a | 8.940 |
| 2 | 2071.155 | 9.238 | −0.0703 | 0.002 | |
| [31], n = 3 | 1 | 2068.713 | 9.436 | n/a | 0.035 |
| 2 | 2068.682 | 9.378 | −0.0192 | exact |
Table 5.
Model parameters for rubidium iodide (MHz) (85Rb127I, n = 10; 87Rb127I, n = 3).
| Model | Be | αe | γe | De1 | SSE | |
|---|---|---|---|---|---|---|
| 85Rb127I | 1 | 984.221 | 3.261 | n/a | n/a | 1.013 |
| 2 | 984.245 | 3.284 | 0.0033 | n/a | 0.237 | |
| 3 | 984.307 | 3.259 | n/a | 289 | 0.644 | |
| 4 | 984.236 | 3.279 | 0.0027 | −81 | 0.113 | |
| 87Rb127I | 1 | 970.681 | 3.204 | n/a | n/a | 0.014 |
| 2 | 970.672 | 3.188 | −0.0056 | n/a | exact |
1 Centrifugal distortion constants have been multiplied by 106.
Table 6.
Model parameters for rubidium bromide (MHz) (85Rb79Br, n = 6; 87Rb79Br, n = 3; 85Rb81Br, n = 3).
Table 6.
Model parameters for rubidium bromide (MHz) (85Rb79Br, n = 6; 87Rb79Br, n = 3; 85Rb81Br, n = 3).
| Model | Be | αe | γe | De1 | SSE | |
|---|---|---|---|---|---|---|
| 85Rb79Br | 1 | 1424.768 | 5.542 | n/a | n/a | 0.432 |
| 2 | 1424.798 | 5.583 | 0.0086 | n/a | 0.041 | |
| 3 | 1424.822 | 5.538 | n/a | −0.79 | 0.268 | |
| 4 | 1424.822 | 5.576 | 0.0076 | −0.40 | 0.005 | |
| 87Rb79Br | 1 | 1409.003 | 5.456 | n/a | n/a | 0.011 |
| 2 | 1409.014 | 5.478 | 0.0072 | n/a | exact | |
| 85Rb81Br | 1 | 1406.546 | 5.450 | n/a | n/a | 0.020 |
| 2 | 1406.562 | 5.479 | 0.0097 | n/a | exact |
1 Centrifugal distortion constants have been multiplied by 106.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Pye, C.C. Interpreting the Microwave Spectra of Diatomic Molecules. Spectrosc. J. 2023, 1, 3-27. https://doi.org/10.3390/spectroscj1010002
AMA Style
Pye CC. Interpreting the Microwave Spectra of Diatomic Molecules. Spectroscopy Journal. 2023; 1(1):3-27. https://doi.org/10.3390/spectroscj1010002
Chicago/Turabian StylePye, Cory C. 2023. "Interpreting the Microwave Spectra of Diatomic Molecules" Spectroscopy Journal 1, no. 1: 3-27. https://doi.org/10.3390/spectroscj1010002