
Abstract
Glioblastoma (GBM) is an incurable brain cancer that lacks effective therapies. Here we show that EAG2 and Kvβ2, which are predominantly expressed by GBM cells at the tumor–brain interface, physically interact to form a potassium channel complex due to a GBM-enriched Kvβ2 isoform. In GBM cells, EAG2 localizes at neuron-contacting regions in a Kvβ2-dependent manner. Genetic knockdown of the EAG2–Kvβ2 complex decreases calcium transients of GBM cells, suppresses tumor growth and invasion and extends the survival of tumor-bearing mice. We engineered a designer peptide to disrupt EAG2–Kvβ2 interaction, thereby mitigating tumor growth in patient-derived xenograft and syngeneic mouse models across GBM subtypes without overt toxicity. Neurons upregulate chemoresistant genes in GBM cells in an EAG2–Kvβ2-dependent manner. The designer peptide targets neuron-associated GBM cells and possesses robust efficacy in treating temozolomide-resistant GBM. Our findings may lead to the next-generation therapeutic agent to benefit patients with GBM.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 digital issues and online access to articles
$119.00 per year
only $9.92 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout








Similar content being viewed by others


Data availability
The spatial expression of EAG2, Kvβ2 and other genes in GBM geometrical regions was derived from the Ivy Glioblastoma Atlas Project (https://glioblastoma.alleninstitute.org). The structure of Kvβ2 is available at the Protein Data Bank with accession code 3eau (https://www.ebi.ac.uk/pdbe/entry/pdb/3eau). IlluminaHiseq RNA-seq data of The Cancer Genome Atlas LGGs and GBMs are available at NCI’s Genomic Data Commons (https://gdc.cancer.gov/about-data/publications/lgggbm_2016). The clinical information and gene expression datasets of CGGA (mRNAseq_693, Illumina HiSeq) are available at the Chinese Glioma Genome Atlas (http://www.cgga.org.cn/index.jsp). Previously published microarray data of TMZ-resistant and -sensitive GBM clones that were reanalyzed here are available at ArrayExpress under accession code E-MTAB-2693 (https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-2693/). Preprocessed transcript data from the GLASS Consortium are available at Synapse (https://www.synapse.org/glass). Bulk RNA-seq data from GBM cell–neuron coculture, and scRNA-seq data from the PDX GBM model with peptide treatment reported in this manuscript, have been deposited in the NCBI Gene Expression Omnibus under accession code GSE231577. All unique materials such as patient-derived cell cultures are freely available and can be obtained by contacting the corresponding author and with a standard material transfer agreement with The Hospital for Sick Children. Data for all figures can be found within the manuscript, in the accompanying source data or from the corresponding author on reasonable request. Source data are provided with this paper.
Code availability
All codes used in this manuscript are freely available at GitHub (https://github.com/l-magnificence/Sequencing-Data-Analysis-Code-for-EAG2-Project).
References
Kotecha, R., Odia, Y., Khosla, A. A. & Ahluwalia, M. S. Key clinical principles in the management of glioblastoma. JCO Oncol. Pract. 19, 180–189 (2023).
Stupp, R. et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 352, 987–996 (2005).
Lee, S. Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 3, 198–210 (2016).
Johnson, B. E. et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 343, 189–193 (2014).
van Thuijl, H. F. et al. Evolution of DNA repair defects during malignant progression of low-grade gliomas after temozolomide treatment. Acta Neuropathol. 129, 597–607 (2015).
Pan, Y. et al. NF1 mutation drives neuronal activity-dependent initiation of optic glioma. Nature 594, 277–282 (2021).
Chen, P. et al. Olfactory sensory experience regulates gliomagenesis via neuronal IGF1. Nature 606, 550–556 (2022).
Magnon, C. et al. Autonomic nerve development contributes to prostate cancer progression. Science 341, 1236361 (2013).
Venkatesh, H. S. et al. Neuronal activity promotes glioma growth through neuroligin-3 secretion. Cell 161, 803–816 (2015).
Venkatesh, H. S. et al. Targeting neuronal activity-regulated neuroligin-3 dependency in high-grade glioma. Nature 549, 533–537 (2017).
Dolma, S. et al. Inhibition of dopamine receptor D4 impedes autophagic flux, proliferation, and survival of glioblastoma stem cells. Cancer Cell 29, 859–873 (2016).
Hayakawa, Y. et al. Nerve growth factor promotes gastric tumorigenesis through aberrant cholinergic signaling. Cancer Cell 31, 21–34 (2017).
Venkataramani, V. et al. Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature 573, 532–538 (2019).
Venkatesh, H. S. et al. Electrical and synaptic integration of glioma into neural circuits. Nature 573, 539–545 (2019).
Curry, R. N. et al. Glioma epileptiform activity and progression are driven by IGSF3-mediated potassium dysregulation. Neuron 111, 682–695 (2023).
Zeng, Q. et al. Synaptic proximity enables NMDAR signalling to promote brain metastasis. Nature 573, 526–531 (2019).
Anastasaki, C., Gao, Y. & Gutmann, D. H. Neurons as stromal drivers of nervous system cancer formation and progression. Dev. Cell 58, 81–93 (2023).
Winkler, F. et al. Cancer neuroscience: state of the field, emerging directions. Cell 186, 1689–1707 (2023).
Osswald, M. et al. Brain tumour cells interconnect to a functional and resistant network. Nature 528, 93–98 (2015).
Yu, K. et al. PIK3CA variants selectively initiate brain hyperactivity during gliomagenesis. Nature 578, 166–171 (2020).
Krishna, S. et al. Glioblastoma remodelling of human neural circuits decreases survival. Nature 617, 599–607 (2023).
Hille, B. Ion Channels of Excitable Membranes 3rd edn (Sinauer, 2001).
Bagal, S. K. et al. Ion channels as therapeutic targets: a drug discovery perspective. J. Med. Chem. 56, 593–624 (2013).
Hutchings, C. J., Colussi, P. & Clark, T. G. Ion channels as therapeutic antibody targets. MAbs 11, 265–296 (2019).
Pongs, O. & Schwarz, J. R. Ancillary subunits associated with voltage-dependent K+ channels. Physiol. Rev. 90, 755–796 (2010).
Liu, X., Chang, Y., Reinhart, P. H., Sontheimer, H. & Chang, Y. Cloning and characterization of glioma BK, a novel BK channel isoform highly expressed in human glioma cells. J. Neurosci. 22, 1840–1849 (2002).
Sales, T. T. et al. Suppression of the Eag1 potassium channel sensitizes glioblastoma cells to injury caused by temozolomide. Oncol. Lett. 12, 2581–2589 (2016).
Venturini, E. et al. Targeting the potassium channel Kv1.3 kills Glioblastoma cells. Neurosignals 25, 26–38 (2017).
Puchalski, R. B. et al. An anatomic transcriptional atlas of human glioblastoma. Science 360,660–663 (2018).
Varn, F. S. et al. Glioma progression is shaped by genetic evolution and microenvironment interactions. Cell 185, 2184–2199 (2022).
Weil, S. et al. Tumor microtubes convey resistance to surgical lesions and chemotherapy in gliomas. Neurol. Oncol. 19, 1316–1326 (2017).
Park, N. I. et al. ASCL1 reorganizes chromatin to direct neuronal fate and suppress tumorigenicity of glioblastoma stem cells. Cell Stem Cell 21, 209–224 (2017).
Hamed, A. A. et al. A brain precursor atlas reveals the acquisition of developmental-like states in adult cerebral tumours. Nat. Commun. 13, 4178 (2022).
Pan, Y. et al. Cortisone dissociates the Shaker family K+ channels from their beta subunits. Nat. Chem. Biol. 4, 708–714 (2008).
Magill, S. T., Choy, W., Nguyen, M. P. & McDermott, M. W. Ommaya reservoir insertion: a technical note. Cureus 12, e7731 (2020).
Uetani, N., Chagnon, M. J., Kennedy, T. E., Iwakura, Y. & Tremblay, M. L. Mammalian motoneuron axon targeting requires receptor protein tyrosine phosphatases sigma and delta. J. Neurosci. 26, 5872–5880 (2006).
Suto, F. et al. Plexin-a4 mediates axon-repulsive activities of both secreted and transmembrane semaphorins and plays roles in nerve fiber guidance. J. Neurosci. 25, 3628–3637 (2005).
Rosenstein, J. M., Krum, J. M. & Ruhrberg, C. VEGF in the nervous system. Organogenesis 6, 107–114 (2010).
McGregor, C. E. & English, A. W. The role of BDNF in peripheral nerve regeneration: activity-dependent treatments and Val66Met. Front. Cell Neurosci. 12, 522 (2018).
Yoon, C. et al. Low-density lipoprotein receptor-related protein 1 (LRP1)-dependent cell signaling promotes axonal regeneration. J. Biol. Chem. 288, 26557–26568 (2013).
Goldshmit, Y. et al. EphA4 blockers promote axonal regeneration and functional recovery following spinal cord injury in mice. PLoS ONE 6, e24636 (2011).
Sanchez-Huertas, C. et al. The +TIP Navigator-1 is an actin-microtubule crosslinker that regulates axonal growth cone motility. J. Cell Biol. https://doi.org/10.1083/jcb.201905199 (2020).
Li, H. et al. Transcription factor MEF2C influences neural stem/progenitor cell differentiation and maturation in vivo. Proc. Natl Acad. Sci. USA 105, 9397–9402 (2008).
Ohkubo, Y., Uchida, A. O., Shin, D., Partanen, J. & Vaccarino, F. M. Fibroblast growth factor receptor 1 is required for the proliferation of hippocampal progenitor cells and for hippocampal growth in mouse. J. Neurosci. 24, 6057–6069 (2004).
Liu, X. et al. Effect of Spp1 on nerve degeneration and regeneration after rat sciatic nerve injury. BMC Neurosci. 18, 30 (2017).
Kim, Y. et al. Perspective of mesenchymal transformation in glioblastoma. Acta Neuropathol. Commun. 9, 50 (2021).
Meyer, M. et al. Single cell-derived clonal analysis of human glioblastoma links functional and genomic heterogeneity. Proc. Natl Acad. Sci. USA 112, 851–856 (2015).
Wang, H. et al. LGALS3 promotes treatment resistance in glioblastoma and is associated with tumor risk and prognosis. Cancer Epidemiol. Biomarkers Prev. 28, 760–769 (2019).
Venkataramani, V. et al. Disconnecting multicellular networks in brain tumours. Nat. Rev. Cancer 22, 481–491 (2022).
Huang, X. et al. Voltage-gated potassium channel EAG2 controls mitotic entry and tumor growth in medulloblastoma via regulating cell volume dynamics. Genes Dev. 26, 1780–1796 (2012).
Monje, M. et al. Roadmap for the emerging field of cancer neuroscience. Cell 181, 219–222 (2020).
Venkataramani, V., Tanev, D. I., Kuner, T., Wick, W. & Winkler, F. Synaptic input to brain tumors: clinical implications. Neurol. Oncol. 23, 23–33 (2021).
Huang, X. et al. EAG2 potassium channel with evolutionarily conserved function as a brain tumor target. Nat. Neurosci. 18, 1236–1246 (2015).
Francisco, M. A. et al. Chloride intracellular channel 1 cooperates with potassium channel EAG2 to promote medulloblastoma growth. J. Exp. Med. https://doi.org/10.1084/jem.20190971 (2020).
Serrano-Heras, G. et al. Involvement of N-methylpurine DNA glycosylase in resistance to temozolomide in patient-derived glioma cells. Sci. Rep. 10, 22185 (2020).
Huang, L. et al. Inhibition of intermedin (adrenomedullin 2) suppresses the growth of glioblastoma and increases the antitumor activity of temozolomide. Mol. Cancer Ther. 20, 284–295 (2021).
Aarts, M. et al. Treatment of ischemic brain damage by perturbing NMDA receptor–PSD-95 protein interactions. Science 298,846–850 (2002).
Cook, D. J., Teves, L. & Tymianski, M. A translational paradigm for the preclinical evaluation of the stroke neuroprotectant Tat-NR2B9c in gyrencephalic nonhuman primates. Sci. Transl. Med. 4, 154ra133 (2012).
Acknowledgements
This work is supported by the SickKids Foundation, Arthur and Sonia Labatt Brain Tumour Research Centre, Garron Family Cancer Centre, Canadian Cancer Society Challenge Grant, Concern Foundation Conquer Cancer Now Award, b.r.a.i.n.child, Sontag Foundation Distinguished Scientist Award, Meagan’s HUG, Natural Sciences and Engineering Research Council Discovery Grant, Ontario Institute for Cancer Research Translational Research Initiative, Early Researcher Award and Canadian Institute of Health Research Project Grants (to X.H.), a Garron Family Cancer Centre Pitblado Discovery Grant (to L.-Y.W. and X.H.), Canadian Institute of Health Research Project Grants PJT-156034 and PJT-156439 and a Natural Sciences and Engineering Research Council grant RGPIN-2017-06665 (to L.-Y.W.). Xin C. is supported by SickKids Restracomp Scholarship. S.B. is supported by an International Postdoc Grant from the Swedish Research Council (Vetenskapsrådet) and Powered by Pablove Research Grant from Pablove Foundation. We thank J. Lathia for sharing GL261 cells, S. O. Kelley for sharing PC9 and H1975 cells and J. Ellis for sharing HEK293T cells, human astrocytes and lentivirus packaging plasmids. We thank P. Paroutis and K. Lau at SickKids Imaging Facility for help with confocal imaging and image analysis. We thank C. Simpson at SickKids SPARC BioCentre Molecular Analysis for assistance with mass spectrometry experiments. We thank the staff at The Centre for Phenogenomic for their help on animal works. We thank W. Wang for technical assistance and Huang laboratory members for comments on the manuscript. Schematics in Figs. 2a, 5c,e, 7a and 8 created with BioRender.com. X.H. is a Catalyst Scholar at The Hospital for Sick Children and Canada Research Chair in Cancer Biophysics.
Author information
Authors and Affiliations
Contributions
X.H. directed the study. Experimental design, data acquisition and data analysis were carried out by W.D., A.F., Xiaodi C., H.L., G.L.B., Xin C., S.B., Y.X., Q.Y., H.Z., T.K., M.S.M., G.J., J.-E.K., G.J., Y.S., T.-H.K., Y.H., S.W., X.L., R.A.M. and L.-Y.W. Reagent contributions were provided by D.S. and P.B.D. W.D. and X.H. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
W.D. and X.H. have filed a PCT patent for the composition of matter and use of designer peptides to treat cancer. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Cancer thanks Justin Lathia, Stephen Robbins and Harald Sontheimer for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 EAG2 and Kvβ2 expression in xenograft GBM tumors, EAG2 localization in GBM cells, EAG2 and Kvβ2 knockdown validation, and tumor growth comparison between control tumor and tumor with knockdown of EAG2, Kvβ2, or both.
a. EAG2, Kvβ2, GFP, and DAPI expression in G411 xenograft tumor. EAG2, Tau, and GFAP expression in G489, G523, and G532 xenograft tumors. b. Representative images and quantification of EAG2 localization in GBM cells with or without contact of astrocytes. n = 24 cells examined over 3 independent experiments. P values, two-sided unpaired t-test. Error bars, mean ± s.e.m. c. Validation of Dox-induced knockdown of EAG2 and Kvβ2. d. Bioluminescence images show tumor burdens of mice bearing G411 tumors before and after Dox treatment. Kaplan-Meier survival curves are shown. P values, log-rank test. All experiments were performed 3 times using biologically independent samples.
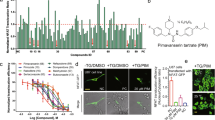
Extended Data Fig. 2 Characterization of peptide treatment on GBM cell growth in vitro and tumor growth in vivo.
a. Co-IP shows EAG2-Kvβ2 interaction in G411 GBM cells treated with TAT, K59-78TAT, K90-114, or K90-114TAT. 3 biologically independent experiments were performed. b. Cell number, microtube number per cell, and microtube length of G489 GBM cells, which are co-cultured with neurons, after TAT, K59-78TAT, K90-114, or K90-114TAT treatment. Sample size (from left to right): n = 31, 42, 31, 43, 50, 52, 33, 41, 78, 146, 116, 49 cells examined over 3 independent experiments. Adjusted P value was calculated by Tukey’s multiple comparisons test (n.s. means not significant, P > 0.05). Error bars, mean ± s.e.m. Cell number and microtube length of K59-78TAT and K90-114TAT treated cells are also shown in Fig. 5b for easier data interpretation. c. Representative images and quantification of tumor areas of G411 xenograft tumors treated for 7 days with K59-78TAT or K90-114TAT. n = 15 samples examined from 5 biologically independent animals. P values, two-sided unpaired t-test. Error bars, mean ± s.e.m. d. Bioluminescence images and survival comparison of G411 GBM-bearing mice treated with K59-78TAT or K90-114TAT at various dosages. P values, log-rank test. Each P value was generated individually by comparing to K59-78TAT. e. Bioluminescence images and survival comparison of G411 GBM-bearing mice treated with TAT, K90-114, or randomized K90-114 TAT. K59-78TAT survival curve is derived from Fig. 5d. P values, log-rank test.
Extended Data Fig. 3 K90-114TAT treatment alters proliferation and apoptosis of GBM tumors.
Representative images showing proliferation and apoptosis of G489 xenograft tumors (pHis3: n = 54, cleaved caspase 3: n = 53) or GL261 syngeneic tumors (pHis3: n = 53 and 48, cleaved caspase 3: n = 54) treated with K59-78TAT or K90-114TAT. STEM121 labels G489 tumor cells. GFP labels GL261 tumor cells. pHis3 labels mitotic cells. cleaved caspase 3 labels apoptotic cells. Samples were evenly and independently collected from 3 animals. P values, two-sided unpaired t-test. Error bars, mean ± s.e.m.
Extended Data Fig. 4 K90-114TAT treatment does not affect mouse body weight, survival, and internal organs.
a. Body weight of K59-78TAT- and K90-114TAT-treated tumor-free NSG mice (n = 9 mice in each group). b. Survival of K59-78TAT- and K90-114TAT-treated tumor-free NSG mice (n = 9 mice in each group). c. Pathological analysis of hearts, kidneys, livers, and lungs harvested from control, K59-78TAT and K90-114TAT-treated tumor-bearing NSG mice (n = 3 mice in each group).
Extended Data Fig. 5 K90-114TAT treatment decreases proliferation and increases apoptosis of TMZ-resistant GBM.
Representative images showing STEM121+ tumor cells, pHis3+ mitotic cells, and cleaved caspase 3+ apoptotic cells in TMZ-resistant xenograft tumors treated with TMZ, K59-78TAT and K90-114TAT. Sample size (from left to right): G411-TMZr: n = 24, 25, 21, 24, 24, 24; G532-TMZr: n = 7, 15, 21, 17, 17, 21. Samples were evenly and independently collected from 3 animals. P values, two-sided unpaired t-test. Error bars, mean ± s.e.m.
Supplementary information
Supplementary Table 1
Sequences of peptides, shRNAs and PCR primers.
Source data
Source Data Fig. 1
Statistical Source Data.
Source Data Fig. 2
Statistical Source Data.
Source Data Fig. 3
Statistical Source Data.
Source Data Fig. 4
Statistical Source Data.
Source Data Fig. 5
Statistical Source Data.
Source Data Fig. 6
Statistical Source Data.
Source Data Fig. 7
Statistical Source Data.
Source Data Extended Data Fig. 1.
Statistical Source Data.
Source Data Extended Data Fig. 2.
Statistical Source Data.
Source Data Extended Data Fig. 3.
Statistical Source Data.
Source Data Extended Data Fig. 4.
Statistical Source Data.
Source Data Extended Data Fig. 5.
Statistical Source Data.
Source Data
Unprocessed immunoblots and gels.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Dong, W., Fekete, A., Chen, X. et al. A designer peptide against the EAG2–Kvβ2 potassium channel targets the interaction of cancer cells and neurons to treat glioblastoma. Nat Cancer 4, 1418–1436 (2023). https://doi.org/10.1038/s43018-023-00626-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s43018-023-00626-8
This article is cited by
-
Pan-cancer ion transport signature reveals functional regulators of glioblastoma aggression
The EMBO Journal (2024)
-
The never-abating excitement for targeted therapies
Nature Cancer (2023)
-
Targeting network circuitry in glioma
Nature Cancer (2023)
